一种制备芳香醛的方法
1.本技术涉及一种制备芳香醛的方法,属于化工催化技术领域。
背景技术:
2.对甲基苯甲醛及其衍生物是合成抗癌药物的前体,属于高端精细化学品,目前通过石油产品对二甲苯的光氯化、有机胺化、盐酸水解等工艺获得。由于生产过程对环境破坏严重已在国内禁止使用,产品从国外进口获取,因而造成价格攀升,给市场注入不稳定因素。虽然甲苯的羰基化路线是潜在替代工艺,然而卤化物等强酸液排放制约了该工艺的规模化工业应用。此外,氯元素的取代残留也抑制了所得产品不能直接用于药物合成。综上,国内亟需开发绿色的对甲基苯甲醛生产工艺,突破现有瓶颈。
3.从含氧、低碳分子构筑芳香化学品是当前研究热点。随着生物质基、煤基等乙醇工业项目的蓬勃发展,将乙醇或其脱氢产物乙醛作为平台分子制备高值化学品(如高碳醇、甲基苯甲醛/醇)有望发展成为化石资源替代路线,符合我国“节能减排、绿色低碳”的能源资源发展战略。其中,温和条件下乙醛催化转化制甲基苯甲醛是极具前瞻性的途径,可替代现阶段从石油产品出发制备甲基苯甲醛的路线,具有重要工业应用价值。然而,在甲基苯甲醛产物中,对甲基苯甲醛的选择性小于5%。此外,乙醛链增长过程中存在复杂的竞争反应,产物分布很宽,高选择性生成特定产物极具挑战。因此开发一条由乙醛高选择性催化转化制备甲基苯甲醛的路线符合我国能源可持续发展的战略需求,可以替代或部分替代石油基路线,保证我国经济社会发展的安全和稳定。
技术实现要素:
4.根据本技术的一个方面,提供了一种制备芳香醛的方法,本技术从化合物a出发,通过催化转化制备对甲基苯甲醛及其衍生物的新路线,并提供该催化路线所需的催化剂,特别强调采用简单的“一锅法”实现该催化反应过程,期望能成为芳烃含氧化合物生产的重要替代路线。
5.根据本技术的第一方面,提供了一种制备芳香醛的方法,所述方法包括:
6.(1)将含有催化剂、有机酸助剂和化合物a的物料i,缩合反应,得到中间产物;
7.(2)将含有所述中间产物的物料ii,脱氢反应,即可得到所述芳香醛;
8.所述化合物a选自具有式i所示结构式的化合物中的任一种:
[0009][0010]
在式i中,r选自甲基、具有式i-1所示结构式的基团中的任一种:
[0011][0012]
在式i-1中,r'选自甲基、乙基、c3的烷基中的任一种;
[0013]
所述催化剂选自具有式i所示结构式中的至少一种:
[0014][0015]
在式i中,r1选自氢、羟基、取代的硅氧基中的至少一种;
[0016]
r2、r3独立地选自c6~c
10
的芳基、c3~c
10
的杂环基、取代的c6~c
10
的芳基、取代的c3~c
10
的杂环基中的任一种。
[0017]
可选地,所述取代的c6~c
10
的芳基中的取代基选自c2~c
10
的烯基、c1~c
10
的烷基、卤素中的任一种;
[0018]
所述取代的c3~c
10
的杂环基中的取代基选自c1~c
10
的烷基;
[0019]
所述取代的硅氧基中的取代基选自c1~c
10
的烷基。
[0020]
可选地,在所述步骤(1)中得到的中间产物为烯醛类化合物。
[0021]
具体地,本技术所采用的醛(化合物a)通过二聚成环生成具有环状结构的烯醛类化合物,然后通过脱氢剂,实现环状结构的烯醛类产品脱氢芳构化生成具有对位取代基的芳香醛。
[0022]
可选地,本技术中的原料为乙醛时,反应式如下:
[0023][0024]
可选地,本技术中的制备芳香醛的方法包括以下步骤:
[0025]
(1)取一定比值的催化剂和有机酸助剂投入到有机溶剂中,搅拌均匀后加入化合物a溶液,于一定温度和气氛下进行缩合反应;
[0026]
(2)反应一定时间后,向步骤(1)中投入一定量的脱氢催化剂加速芳构化反应速率;
[0027]
(3)反应结束后,减压蒸馏得到芳香产物,气相色谱分析产物分布,外标工作曲线确定产物量。
[0028]
其中:步骤(1)中,所述催化剂为吡咯烷的衍生物,其具有支链,可以是r构型和s构型,优选r构型;支链上具有烷基、杂环、或芳环基团;烷基可以为甲基、乙基、丙基等;杂环为吡咯、吡啶、咪唑等;芳环上可以含有-h、-ch3、-ch=ch2、-c2h5、
ꢀ‑
och3、cf3、f、br等烷基、烯基、卤素官能团,但不仅限于这些官能团;官能团位置可以是邻位、间位或对位(如下式所示);
[0029][0030]
步骤(1)中,所述催化剂为吡咯烷的衍生物,其支链上还可以具有空间位阻基团,如长链烷基、苯环、甲基硅氧、乙基硅氧、异丙基硅氧等基团;烷基硅氧基团的烷基基团不仅限于低碳烷基,还可以是苯环、杂环等;烷基数目可以是一个、二个或三个,优选含三个烷基取代的硅氧基团,但更优选包含3个甲基取代的硅氧基团,如三甲基硅氧;
[0031]
可选地,所述取代的硅氧基中的取代基选自c1~c
10
的烷基。
[0032]
可选地,所述物料i中还包括有机溶剂;
[0033]
优选地,所述有机溶剂选自三氯甲烷、甲苯、乙腈、乙醇、n,n-二甲基甲酰胺中的至少一种;
[0034]
优选地,所述化合物a在所述物料i中的浓度为0.001~1mol/l。
[0035]
可选地,所述化合物a在所述物料i中的浓度上限独立地选自1、0.8、0.6、0.4、 0.2、0.05、0.01、0.005,下限独立地选自0.001、0.8、0.6、0.4、0.2、0.05、0.01、0.005。
[0036]
优选地,所述化合物a在所述物料i中的浓度为0.1~0.3mol/l。
[0037]
可选地,所述有机酸助剂选自苯酚类化合物、羧酸类化合物中的至少一种。
[0038]
可选地,所述苯酚类化合物选自苯酚、对硝基苯酚、对甲基苯酚中的至少一种;
[0039]
所述羧酸类化合物选自苯甲酸、乙酸中的至少一种。
[0040]
可选地,在所述步骤(1)中,所述催化剂和有机酸助剂的摩尔比为1:5~1:0.0001;
[0041]
所述化合物a和所述催化剂的摩尔比为100:1~1:1,优选20:1~5:1,更优选 20:1~10:1。
[0042]
可选地,在所述步骤(1)中,所述催化剂和有机酸助剂的摩尔比上限独立地选自 1:5、1:4、1:3、1:2、1:1、1:0.1、1:0.01、1:0.001、1:0.0005,下限独立地选自1:0.0001、1:4、1:3、1:2、1:1、1:0.1、1:0.01、1:0.001、1:0.0005。
[0043]
可选地,所述化合物a和所述催化剂的摩尔比上限独立地选自100:1、90:1、 80:1、70:1、60:1、50:1、40:1、30:1、20:1、10:1,下限独立地选自1:1、 90:1、80:1、70:1、60:1、50:1、40:1、30:1、20:1、10:1。
[0044]
可选地,在所述步骤(1)中,所述催化剂和有机酸助剂的摩尔比为1:2~1:0.3。
[0045]
可选地,在所述步骤(1)中,所述催化剂和有机酸助剂的摩尔比上限独立地选自 1:2、1:1、1:0.5,下限独立地选自1:0.3、1:1、1:0.5。
[0046]
可选地,在所述步骤(1)中,所述缩合反应的条件为:温度为-20-100℃;时间为
0.01~100h;
[0047]
在所述步骤(2)中,所述脱氢反应的条件为:温度为-20-100℃;时间为0.01~100 h。
[0048]
可选地,在所述步骤(1)中,所述缩合反应的温度上限独立地选自100℃、90℃、 80℃、70℃、60℃、50℃、40℃、30℃、20℃、10℃,下限独立地选自-20℃、0℃、 90℃、80℃、70℃、60℃、50℃、40℃、30℃、20℃、10℃。
[0049]
可选地,在所述步骤(1)中,所述缩合反应的时间上限独立地选自100h、90h、 80h、70h、60h、50h、24h、20h、16h、12h、8h、4h,下限独立地选自0.01h、 90h、80h、70h、60h、50h、24h、20h、16h、12h、8h、4h。
[0050]
可选地,在所述步骤(2)中,所述脱氢反应的温度上限独立地选自100℃、90℃、80℃、70℃、60℃、50℃、40℃、30℃、20℃、10℃,下限独立地选自-20℃、0℃、 90℃、80℃、70℃、60℃、50℃、40℃、30℃、20℃、10℃。
[0051]
可选地,在所述步骤(2)中,所述脱氢反应的时间上限独立地选自100h、90h、 80h、70h、60h、50h、24h、20h、16h、12h、8h、4h,下限独立地选自0.01h、90h、80h、70h、60h、50h、24h、20h、16h、12h、8h、4h。
[0052]
可选地,在所述步骤(1)中,缩合反应在一定气氛下进行,气氛可以是空气、氩气、氮气、氧气、氢气等气体的一种或多种组合(含氢气和氧气的组合除外),但不仅限于这些气氛;优选空气、氮气或氩气,更优选空气;可以是常压或高压,优选常压。可选地,在所述步骤(2)中,在所述物料ii中,还包括脱氢剂;
[0053]
所述脱氢剂选自二氧化锰、过氧化氢、叔丁基过氧化氢、苯醌、苯醌衍生物中的至少一种;
[0054]
优选地,所述苯醌衍生物选自2,3-二氯-5,6-二氰基苯琨(ddq),四氯苯醌、四氟苯醌、四溴苯醌中的至少一种。
[0055]
可选地,所述脱氢剂与所述化合物a的摩尔比为4∶1~40∶1,优选4∶1~20∶1。
[0056]
可选地,所述脱氢剂与所述化合物a的摩尔比上限独立地选自40∶1、30∶1、20∶ 1、10∶1、5∶1、1∶1,下限独立地选自40∶1、30∶1、20∶1、10∶1、5∶1、1∶1。
[0057]
可选地,本技术通过减压精馏收集馏分,气相色谱确定产物分布,获得产物选择性;外标工作曲线确定产量和收率。所得芳香醛主要为对甲基苯甲醛。对甲基苯甲醛及其前体选择性为90.5%。
[0058]
本技术中,c1~c
10
指所包含的碳原子数。
[0059]
本技术中,“烷基”是由烷烃化合物分子上失去任意一个氢原子所形成的基团。所述烷烃化合物包括直链烷烃、支链烷烃、环烷烃、带有支链的环烷烃。
[0060]
本技术中,“烯基”是由烯烃化合物分子上失去任意一个氢原子所形成的基团。所述烯烃化合物包括直链烯烃、支链烯烃、环烯烃、带有支链的环烯烃。
[0061]
本技术能产生的有益效果包括:
[0062]
与传统的对二甲苯光氯化、有机胺化、酸解等路线相比,该反应路线具有原子经济性高、环境友好、气液易于分离等优点,是一条创新的绿色对甲基苯甲醛及其衍生物的生产路线。该反应路线的反应温度为0-60℃,对甲基苯甲醛及其前体的总选择性高达90.5%,而邻甲基苯甲醛选择性仅为3.3%,总芳香醛选择性为83.8%,具有良好的工业应用前景。该
路线生成一定量的氢气,且产物中不含有氯元素(作为取代基)残留,可直接用于医药化合物的生产。此外,该路线还生成了部分邻甲基苯甲醛,其同样可以作为高附加值产品,用于药物、试剂等的合成,部分替代石油基产品。产品可以通过减压蒸馏与催化剂和有机酸分离,然后通过精馏进一步除去溶剂,得到高纯度产品,所以本发明所提供的反应路径具有极大地工业化应用前景。所以本专利的创新为反应路径创新。
附图说明
[0063]
图1是实施例2中催化剂ii上的产物分布图谱。
具体实施方式
[0064]
下面结合实施例详述本技术,但本技术并不局限于这些实施例。
[0065]
如无特别说明,本技术的实施例中的原料均通过商业途径购买。
[0066]
对反应物和产物的分析,以及转化率和选择性的计算,做如下描述:
[0067]
采用agilent 7900b气相色谱分析反应后的溶液,得到产物的峰面积和未转化的原料峰面积,通过摩尔校正因子,采用校正面积归一法计算转化率和产物的选择性。由于涉及到产物分子中碳数数目的变化,所以计算所得的转化率和选择性均是基于无摩尔量纲的摩尔碳数。
[0068]
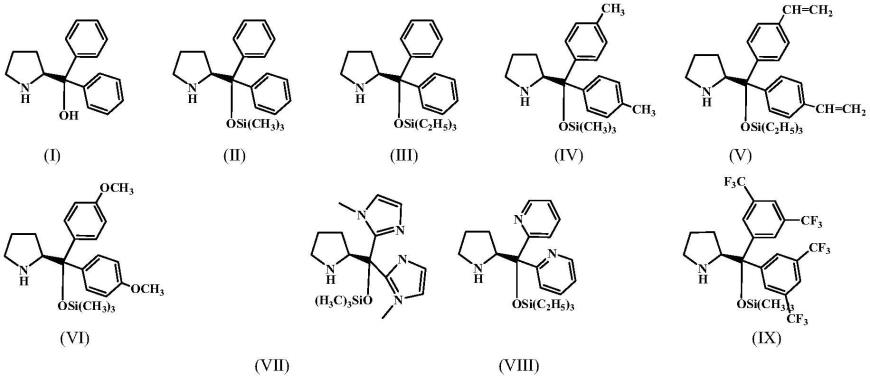
实施例中使用的催化剂结构及编号如下:
[0069][0070]
实施例(1)
[0071]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化剂i和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢10mmol,继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0072]
产物分析结果如下:乙醛转化率为《3%,对甲基苯甲醛选择性为70.8%,产品纯度可为》99%。
[0073]
实施例(2)
[0074]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂二氯甲烷、3mmol有机酸对硝基苯酚、1 mmol催化剂ii和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢10mmol,继续搅拌
2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0075]
图1是实施例2中催化剂ii上的产物分布图谱,产物分析结果如下:乙醛转化率为》95%,对甲基苯甲醛选择性为80.1%,对甲基苯甲醛及其前体总选择性为89.5%,其中对甲基苯甲醛的产品纯度可为》99%。
[0076]
实施例(3)
[0077]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化剂iii和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢10mmol,继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0078]
产物分析结果如下:产物分析结果如下:乙醛转化率为》95%,对甲基苯甲醛选择性为80.0%,对甲基苯甲醛及其前体总选择性为89.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0079]
实施例(4)
[0080]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、0.5mmol有机酸对硝基苯酚、 1mmol催化剂iv和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0081]
产物分析结果如下:产物分析结果如下:乙醛转化率为》95%,对甲基苯甲醛选择性为79.0%,对甲基苯甲醛及其前体总选择性为88.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0082]
实施例(5)
[0083]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化剂v和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0084]
产物分析结果如下:产物分析结果如下:乙醛转化率为》95%,对甲基苯甲醛选择性为79.0%,对甲基苯甲醛及其前体总选择性为88.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0085]
实施例(6)
[0086]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、1mmol催化剂vi和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0087]
产物分析结果如下:产物分析结果如下:乙醛转化率为》95%,对甲基苯甲醛选择性为77.0%,对甲基苯甲醛及其前体总选择性为89.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0088]
实施例(7)
[0089]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化
剂vii和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0090]
产物分析结果如下:乙醛转化率为96.1%,对甲基苯甲醛选择性为70.0%,对甲基苯甲醛及其前体总选择性为79.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0091]
实施例(8)
[0092]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化剂viii和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0093]
产物分析结果如下:乙醛转化率为95.0%,对甲基苯甲醛选择性为60.0%,对甲基苯甲醛及其前体总选择性为79.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0094]
实施例(9)
[0095]
以乙醛为原料,在液相高压反应釜中开展乙醛偶联-芳构化反应活性测试。反应条件如下:在75ml高压釜中加入30ml溶剂(二氯甲烷)、3mmol有机酸对硝基苯酚、 1mmol催化剂ix和乙醛(10mmol)。在常压下,于20℃,反应2h后,投入叔丁基过氧化氢(10mmol),继续搅拌2h。待反应结束后,反应原料及产物利用离线色谱分析,产物结构通过质谱确定。
[0096]
产物分析结果如下:乙醛转化率为98.0%,对甲基苯甲醛选择性为80.0%,对甲基苯甲醛及其前体总选择性为89.0%,其中对甲基苯甲醛的产品纯度可为》99%。
[0097]
实施例(10)
[0098]
反应条件同实施例(2),但采用不同摩尔量的ch3cho作为反应物(ch3cho浓度为0.001mol/l-1mol/l)。
[0099]
产物分析结果如下表1所示:
[0100]
表1
[0101][0102][0103]
实施例(11)
[0104]
反应条件同实施例(2),但采用采用不同摩尔比的有机酸对硝基苯酚作为助催化剂。
[0105]
产物分析结果如下表2所示:
[0106]
表2
[0107][0108]
实施例(12)
[0109]
反应条件同实施例(2),但采用不同温度进行反应,反应时间固定为是100h。
[0110]
产物分析结果如下表3所示:
[0111]
表3
[0112][0113][0114]
实施例(13)
[0115]
反应条件同实施例(2),但采用不同量的叔丁基过氧化氢作为脱氢试剂。
[0116]
产物分析结果如下表4所示:
[0117]
表4
[0118][0119]
实施例(14)
[0120]
反应条件同实施例(2),但采用等摩尔量的其它有机酸替代对硝基苯酚。
[0121]
产物分析结果如下表5所示:
[0122]
表5
[0123]
有机酸(℃)转化率(%)对甲基苯甲醛选择性(%)苯酚86.933.7对甲基苯酚77.423.2对羟基苯甲酸82.333.3对硝基苯甲酸50.54.4对硝基苯酚》9980.5
[0124] 实施例(15)
[0125]
反应条件同实施例(2),但采用等体积的其它溶剂代替二氯甲烷。
[0126]
产物分析结果如下表6所示:
[0127]
表6
[0128]
溶剂(℃)转化率(%)对甲基苯甲醛选择性(%)甲苯90.579.4乙腈77.421.5乙醇27.630.8三氯甲烷》9980.5
[0129]
实施例(16)
[0130]
反应条件同实施例(2),但加入ddq作为脱氢剂替代叔丁基过氧化氢。
[0131]
产物分析结果如下:乙醛转化率为94.3%,对甲基苯甲醛选择性为79.2%,产品纯度可为》96%。
[0132]
实施例(17)
[0133]
反应条件同实施例(2),但加入5mmol丁烯醛作为反应原料。
[0134]
产物分析结果如下:丁烯醛转化率为90.3%,对甲基苯甲醛选择性为79.1%,产品纯度可为》96%。
[0135]
实施例(18)
[0136]
反应条件同实施例(2),但加入5mmol 2-戊烯醛作为反应原料。
[0137]
产物分析结果如下:2-戊烯醛转化率为85.0%,3-甲基-4-乙基-苯甲醛选择性为 90.0%,产品纯度可为》96%。
[0138]
实施例(19)
[0139]
反应条件同实施例(2),但加入5mmol 2-辛烯醛作为反应原料。
[0140]
产物分析结果如下:2-辛烯醛转化率为81.0%,3-丁基-4-戊基-苯甲醛选择性为95.5%,产品纯度可为》96%。
[0141]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1