无毒CAS9酶及其应用的制作方法
无毒cas9酶及其应用
交叉引用
1.本技术要求于2019年1月7日提交的美国临时申请62/789,347;于2019年3月25日提交的美国临时申请62/823,477;于2019年3月26日提交的美国临时申请62/824,164和于2019年5月31日提交的美国临时申请62/855,612的优先权,通过引用将其全部内容并入本文。序列表
2.本技术包含序列表,所述序列表已以ascii格式电子提交,并通过引用整体并入本文。创建于2020年1月3日的所述ascii副本被命名为55190
‑
701_601_sl.txt,并且大小是236,317字节。
背景技术:
3.核酸的靶向编辑是研究遗传功能以及治疗和改善遗传障碍和疾病的症状的一种非常有前景的方法。最显著的靶标特异性遗传修饰方法涉及锌指核酸酶(zfn)、转录激活因子样效应核酸酶(talen)和rna指导的dna内切核酸酶cas的工程改造和使用。通过非同源末端连接(nhej)修复机制在靶核酸处引入突变(例如缺失和插入)的频率限制了基因靶向和编辑在治疗剂开发中的应用。
技术实现要素:
4.在此处的本公开内容部分地概括在本文公开的权利要求书中。本文公开了一种方法,其包括将第一载体引入多个细胞中,其中所述第一载体编码融合蛋白复合物,所述融合蛋白复合物包含与外切核酸酶融合的cas9核酸酶;其中包含所述载体的所述多个细胞的活力是包含编码cas9核酸酶的第二载体的第二多个细胞的活力的至少1.5倍;其中所述第二多个细胞是用所述第二载体转染的k562细胞。第一载体可以编码与外切核酸酶融合的cas9和grna。外切核酸酶可以选自mre11、exol、exoiii、exovii、exot、dna2、ctip、trex1、trex2、apollo、rece、recj、t5、lexo、recbcd和mungbean。可以将供体多核苷酸引入第一多个细胞中。所述方法可以包括通过与外切核酸酶融合的所述cas9对基因的异常基因座进行编辑。供体多核苷酸可包含还含有所述基因的功能性基因座的整合盒。活力可以通过刃天青测定来测量。外切核酸酶可以是exoi。异常基因座可以是hbb基因的异常基因座。供体多核苷酸可以编码所述hbb基因的功能性基因座。融合蛋白复合物可以编码至少一个核定位信号(nls)。编码融合蛋白复合物的第一载体可以与seq id no:2
‑
18中的任一个具有至少80%序列同一性。第一载体可以通过电穿孔递送。供体多核苷酸可包含位于紧邻切割位点的3’末端的突变前间区序列邻近基序(pam)序列,其中所述突变pam序列包含5
’‑
ncg
‑3’
或5
’‑
ngc
‑3’
。融合蛋白复合物不能切割所述突变pam序列。供体多核苷酸可以是单链dna。供体多核苷酸可以是双链dna。
5.本文公开了多肽,其包含第一功能片段、包含cas核酸酶的第二功能片段和接头肽,其中所述第一功能片段与所述接头肽的第一末端偶联,并且所述第二功能片段与所述
接头肽的第二末端偶联;并且当将包含所述多肽和核糖核酸(rna)分子的第一复合物施用于第一多个细胞时,与将包含cas9核酸酶和所述rna分子的第二复合物施用于第二多个细胞时在所述第二多个细胞中观察到的毒性相比,在所述第一多个细胞中观察到降低的毒性。所述第一功能片段可包含外切核酸酶,其中所述外切核酸酶选自mre11、exol、exoiii、exovii、exot、dna2、ctip、trex1、trex2、apollo、rece、recj、t5、lexo、recbcd和mungbean。所述rna分子可以是指导rna。所述外切核酸酶可以是人exo1酶。人exo1酶的n末端可偶联至所述接头的所述c末端,所述接头偶联至所述cas核酸酶的所述c末端。人exo1酶可包含seq id no:1。人exo1酶可包含与seq id no:1具有80%序列同一性的片段。人exo1酶可包含与seq id no:1具有90%序列同一性的片段。人exo1酶可包含与seq id no:1具有95%序列同一性的片段。所述第二功能片段可包含cas9酶。cas9酶可包含n末端核定位序列(nls)和c末端nls。cas9酶可包含n末端核定位序列(nls)。cas9酶可包含c末端核定位序列(nls)。接头肽可以选自fl2x、sla2x、ap5x、fl1x、sla1x。接头肽可以是sla2x。肽可包含5至200个氨基酸。降低的毒性可以通过测量试卤灵积累来量化。与施用所述第二复合物之后的所述第二多个细胞相比,在施用所述第一复合物之后,第一多个细胞可以具有至少两倍数量的活细胞,其中活细胞的数量通过试卤灵测定进行量化。当与施用第二复合物后的第二多个细胞相比时,在施用第一复合物后,第一多个细胞具有至少两倍所述数量的hdr编辑细胞,如通过细胞hdr测定所定量的。细胞hdr测定可以包括ihc、qpcr或深度测序。
6.本文公开了一种编码上述多肽和rna分子的多核苷酸。接头肽的第一末端可以是3’末端,接头肽的第二末端可以是5’末端。所述接头肽的第一末端可以是5’末端,所述接头肽的第二末端可以是3’末端。所述rna分子可以是指导rna(grna)。所述多核苷酸可包含同源定向修复(hdr)模板。所述grna可以选自表2中所列的序列。所述hdr模板可以是单链dna。所述hdr模板可以是双链dna。所述多核苷酸可以配制在脂质体中。所述脂质体可包含聚乙二醇(peg)、细胞穿透肽、配体、适体、抗体或其组合。
7.本文公开了一种包含上述多肽的核苷酸序列的载体。所述载体可包含启动子。所述启动子可以是cmv或cag启动子。所述载体可以选自逆转录病毒载体、腺病毒载体、慢病毒载体、疱疹病毒载体和腺伴随病毒载体。所述载体可以是腺伴随病毒载体。本文公开了包含上述载体的病毒样颗粒(vlp)。本文公开了一种试剂盒,其包含在相容的药用赋形剂中配制的上述多肽、带有施用说明的插入物、试剂。
8.本文公开了一种试剂盒,其包含在相容的药用赋形剂中配制的上述多核苷酸、带有施用说明的插入物、试剂。
9.本文公开了一种试剂盒,其包含在相容的药用赋形剂中配制的上述载体、带有施用说明的插入物、试剂。
10.本文公开了一种在细胞中诱导dna同源重组的方法,所述方法包括使dna与上述多肽接触。
11.本文公开了一种在体外或离体细胞中诱导hdr的方法,所述方法包括将上述多核苷酸递送到细胞中。细胞可以是人细胞、非人哺乳动物细胞、干细胞、非哺乳动物细胞、无脊椎动物细胞、植物细胞或单真核生物。
12.本发明公开了一种方法,所述方法包括:将第一多个细胞与上述多核苷酸接触,将第二多个细胞与编码野生型cas9酶的第二多核苷酸接触;以及在预期的基因座处诱导位点
特异性切割,随后在所述第一多个细胞和所述第二多个细胞中进行hdr;以及与所述第二多个细胞相比,在所述第一多个细胞中多回收至少30%
‑
90%的细胞。所述方法可以进一步包括通过测量在所述第一多个细胞和所述第二多个细胞中产生的试卤灵的量来测量细胞活力。当与所述第二多个细胞相比时,所述第一多个细胞可以具有2
‑
5倍量的活细胞,如通过试卤灵测定量化的。所述第一多个细胞和所述第二多个细胞可以包括人细胞、非人哺乳动物细胞、干细胞、非哺乳动物细胞、无脊椎动物细胞、植物细胞或单真核生物。人细胞可以是t细胞、b细胞、树突细胞、自然杀伤细胞、巨噬细胞、中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞、造血祖细胞、造血干细胞(hsc)、红细胞、血干细胞、内胚层干细胞、内胚层祖细胞、内胚层前体细胞、分化的内胚层细胞、间充质干细胞(msc)、间充质祖细胞、间充质前体细胞,或分化的间充质细胞。分化的内胚层细胞可以是肝细胞祖细胞、胰腺祖细胞、肺祖细胞或气管祖细胞。分化的间充质细胞可以是骨细胞、软骨细胞、肌肉细胞、脂肪细胞、基质细胞、成纤维细胞或真皮细胞。
13.本文公开了一种用于治疗受试者的单基因遗传障碍的方法,所述方法包括:培养从所述受试者获得的多个原代细胞;将上述多核苷酸施用于所述多个原代细胞,其中grna被配置为识别导致所述障碍的基因的基因座,并且hdr模板被配置为提供所述基因的功能序列;并且在基因座处诱导位点特异性切割,然后进行hdr,其中所述基因的功能序列插入在所述基因座处。所述方法可以进一步包括选择原代细胞,在所述原代细胞中,所述基因的所述功能序列插入在所述基因座处;并将选定的原代细胞重新引入受试者。受试者可以是哺乳动物。哺乳动物可以是人。多个原代细胞可以选自包括以下的组:t细胞、b细胞、树突细胞、自然杀伤细胞、自然杀伤细胞、巨噬细胞、中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞、造血祖细胞、造血干细胞(hsc)、红细胞、血干细胞、内胚层干细胞、内胚层祖细胞、内胚层前体细胞、分化的内胚层细胞、间充质干细胞(msc)、间充质祖细胞、间充质前体细胞、分化的间充质细胞、肝细胞祖细胞、胰腺祖细胞、肺祖细胞、气管祖细胞、骨细胞、软骨细胞、肌肉细胞、脂肪细胞、基质细胞、成纤维细胞和真皮细胞。引起所述单基因遗传障碍的基因可选自表3。
14.本文公开了一种用于治疗受试者的由异常hbb基因引起的镰状细胞性贫血的方法,所述方法包括:培养从所述受试者获得的多个原代细胞;将上述多核苷酸施用于所述多个原代细胞,其中grna被配置为识别导致所述障碍的所述hbb基因的基因座,并且hdr模板被配置为提供所述hbb基因的功能序列;并且在基因座处诱导位点特异性切割,然后进行hdr,其中所述hbb基因的功能序列插入在所述基因座处。所述方法可以进一步包括选择原代细胞,在所述原代细胞中,所述hbb基因的所述功能序列插入在所述基因座处;并将所述选定的原代细胞重新引入所述受试者。受试者可以是哺乳动物。哺乳动物可以是人。原代细胞可以是造血干细胞。原代细胞可以是cd34+造血干细胞。原代细胞可以是cd34+造血干细胞。载体可包含质粒px330。细胞可以是cd34+造血干细胞。
15.本文公开了一种用于治疗受试者的由异常hbb基因引起的镰状细胞性贫血的方法,所述方法包括:培养从所述受试者获得的多个原代细胞;将上述多核苷酸施用于所述多个原代细胞,其中grna被配置为识别导致所述障碍的所述hbb基因的基因座,并且hdr模板被配置为提供所述hbb基因的功能序列;并且在基因座处诱导位点特异性切割,然后进行hdr,其中所述hbb基因的功能序列插入在所述基因座处。所述方法可以进一步包括选择原
代细胞,在所述原代细胞中,所述hbb基因的所述功能序列插入在所述基因座处;并将选定的原代细胞重新引入受试者。受试者可以是哺乳动物。哺乳动物可以是人。原代细胞可以是cd34+造血干细胞。
16.本发明公开了一种方法,所述方法包括:使第一多个细胞与包含上述多核苷酸和rna分子的第一复合物接触;在所述第一多个细胞中诱导位点特异性切割,然后进行hdr,其中通过细胞hdr测定定量的hdr编辑的第一多个细胞的细胞百分比比与包含编码野生型cas9酶的多核苷酸和所述rna分子的第二复合物接触的第二多个细胞的细胞百分比高至少两倍。细胞hdr测定可以包括ihc。细胞hdr测定可以包括qpcr。细胞hdr测定可以包括核酸测序。援引并入
17.本说明书中提到的所有出版物、专利和专利申请均通过引用并入本文,其程度如同每个单独的出版物、专利或专利申请被明确地并单独地指出通过引用的方式并入一样。
附图说明
18.本专利或申请文件包含至少一个彩色附图。应请求并且支付必要的费用后,具有一个或多个彩色附图的本专利或专利申请公开的副本将由专利局提供。
19.通过参考以下阐述其中利用了本公开的原理的说明性实施方案的详细描述以及附图,可以理解本公开的特征和优点,在附图中:
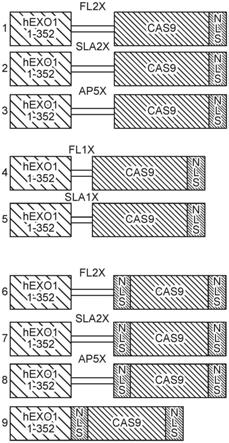
20.图1显示了包含通过不同接头连接在一起的hexo1酶和cas9酶的融合蛋白的实施方案。
21.图2示出了预期靶位点和hdr模板的实施方案。
22.图3显示了进行刃天青还原测定的实施方案。1
‑
8列分别对应于图1中描述的cas9
‑
hr融合蛋白1
‑
8。
23.图4显示了在嘌呤霉素选择之前,用rnp质粒、gfp质粒和对照质粒转染的细胞的试卤灵荧光的归一化倍数变化。
24.图5显示了用野生型cas9酶质粒转染的细胞的试卤灵荧光的归一化倍数变化,所述野生型cas9酶质粒用二甲亚砜(dmso)或pifithrin
‑
α(pft
‑
α)处理。
25.图6a显示了预期靶位点的实施方案,所述靶位点具有三个grna序列(g1、g2和g3;分别为seq id no:21、seq id no:22、seq id no:23)被设计成靶向hbb基因的外显子1。
26.图6b显示了用rnp质粒转染的细胞的试卤灵荧光的归一化倍数变化,所述rnp质粒具有设计为靶向hbb基因的外显子1的三个grna序列(g1、g2和g3;分别为seq id no:21、seq id no:22、seq id no:23)。
27.图6c显示了cas9 hbb
‑
g3反向桑格序列迹线(seq id no:161)。
28.图7显示了进行刃天青还原测定的实施方案。1
‑
9列分别对应于图1中描述的融合蛋白的cas9
‑
hr融合蛋白1
‑
9。
29.图8显示了用具有不同grna序列的rnp质粒、gfp质粒和两种不同对照质粒(针对对照细胞)转染的细胞的试卤灵荧光的归一化倍数变化。
30.图9a
‑
b显示了用rnp质粒转染的细胞的试卤灵荧光的归一化倍数变化。图9a显示
了靶向hbb基因的外显子1的g2(seq id no:22)和g3(seq id no:23)grna。图9b显示了与具有未修饰的cas9和g2 grna的rnp质粒相比,具有第七融合蛋白(图1)和g3 grna的rnp质粒具有更小的细胞毒性。
31.图10显示了用靶向hbb基因的外显子1的不同rnp质粒转染的细胞的试卤灵荧光的归一化倍数变化。
32.图11a是质粒px330的示意图,其包含用于哺乳动物cas9表达的组成型启动子,以及驱动的grna表达的u6启动子。
33.图11b是其中接种细胞并在生长两天后量化细胞毒性的实验设置的示例。
34.图11c是显示如图11b实验设置中所示的a549细胞中降低的细胞毒性的图和该图上方描绘的12号染色体上的grna靶向基因间区域的图。
35.图11d是显示用α
‑
pifithrin(10微摩尔)处理降低a549细胞中cas9诱导的细胞毒性的图。
36.图12a是嘌呤霉素抗性修复模板(rt)的图。
37.图12b显示了用于量化a549细胞中hexo
‑
cas9融合的hdr和indel率的方法。
38.图12c是描绘通过刃天青测定测试的各种构建体的毒性的图。
39.图12d描绘了刃天青测定的方法。
40.图12e描绘了通过puro
‑
rt成功整合的细胞基因组区域。
41.图12f是用具有g2或g3 rna的cas9
‑
hr8(8)或cas9(nt)转染的k562细胞在嘌呤霉素处理三天后的存活率图。
42.图12g是图12e中描绘的引物的扩增产物的琼脂糖凝胶,显示使用cas9
‑
hr8(图1的融合蛋白8)和在基因组中具有grna g2或g3的cas9(nt)的修复模板的稳定整合。
43.图13a显示了基因组区域(包括hbb的前两个外显子,其被靶向以编辑人血红蛋白β(hbb)基因)以及描绘来自a549细胞中hbb grna指导物的毒性筛选数据的图。
44.图13b显示了在hbb
‑
g3处理的a549细胞(seq id no:161)中hbb基因组区域的桑格测序。
45.图13c是野生型hbb序列(seq id no:162)和ssrt
‑
g3序列(seq id no:163)的图,该ssrt
‑
g3序列向镰状细胞(e6v)引入了导致ecori位点的错义突变和四个沉默错配突变(在单链修复模板ssrt g3上的粗体核苷酸a、a、a和g),其中hbb
‑
g3 grna由上面的条突出显示。突变被设计为防止成功修复后grna结合。
46.图13d描绘了hbb编辑实验,其中k562细胞或a549细胞用cas9+ssrt
‑
g3、cas9
‑
hr1
‑
9+ssrt
‑
g3或单独的ssrt
‑
g3进行电穿孔。
47.图14图示了通过用hbb
‑
g3 grna和cas9
‑
hr融合蛋白4和5(如图1所示)转染a549细胞确定的两种转染方法lipofectamine和磷酸钙(calphos)的毒性评估。
48.图15图示了通过用图13a的hbb修复模板转染a549细胞进行的毒性评估。在转染后第2天测量刃天青水平。
49.图16a显示了图1的cas9
‑
hr融合蛋白8的ecori消化测定的琼脂糖凝胶,该融合蛋白将hbb修复模板整合到k562细胞的基因组中。箭头表示ecori消化的产物。在仅cas9(nt)、ssrt和con(无cas9)的泳道中没有ecori消化的产物。
50.图16b显示了图1的cas9
‑
hr融合蛋白4、5、6、7和8的ecori消化测定的琼脂糖凝胶,
该融合蛋白将hbb修复模板整合到k562细胞的基因组中。箭头表示ecori消化的产物。nt和con泳道中没有ecori消化的产物。
51.图16c显示了图1的cas9
‑
hr融合蛋白4、5、6、7和8,仅cas9(nt)和con(无cas9)的蛋白质印迹。箭头表示在cas9
‑
hr融合蛋白和nt泳道中检测到cas9。
52.图16d显示了cas9
‑
hr3在大肠杆菌中的成功表达和纯化。也显示了cas9的成功表达和纯化(泳道8
‑
14)有助于比较。
53.图16e显示了来自图16c的相同转染细胞的免疫组织化学(ihc)。箭头表示cas9
‑
hr融合物和cas9定位于细胞核。
54.图17a图示了完整h2b敲入实验的构建体。
55.图17b图示了由图1的cas
‑
hr融合蛋白4、5、6和8,仅cas9(nt)和con(无cas9)在上皮肺癌细胞系中诱导的细胞毒性的p53依赖性降低。a549细胞对p53活性呈阳性,而h1299细胞对p53活性呈阴性。由归一化刃天青水平(y轴)确定的毒性表明,h1299细胞中p53的缺失产生较低的细胞毒性。
56.图17c图示了在k562细胞中对如图17a所示的h2b的成功gfp标记的评估。箭头表示用gfp成功标记h2b,如在细胞核中检测到gfp所示。
57.图18a图示了仅有cas9的模型和cas9
‑
hr模型之间的示意性差异。外切核酸酶结构域的存在从根本上改变了预测的体外切割模式。exo1对磷酸化的5’末端相对非磷酸化具有显著的偏好。因此,可以预期,当使用pcr产物或其他缺乏5
’‑
磷酸化末端的dna片段时,通过cas9的内切核酸酶切割最初可以占主导地位,而切割后这两个片段各自可以具有5
’‑
磷酸化末端,这通过hexo1导致快速降解。
58.图18b图示了基于图18a的示例性消化模式。仅cas9
‑
hr3+grna和cas9
‑
hr3才能产生证明成功体外核酸酶活性的消化产物。此外,尽管hexo1强烈偏好磷酸化的5’末端,但hexo1仍然可以结合和切除未磷酸化的5’末端,因此与cas9相比,在没有grna情况下有少量降解。
59.图18c图示了图18a和18b的实际琼脂糖示例。泳道1和2显示cas9
‑
hr3靶向hbb
‑
g1或hbb
‑
g3,泳道3和4显示cas9(nt)靶向hbb
‑
g1或hbb
‑
g3,泳道5是未处理的dna。
60.图18d图示了与图18c类似的实验,差异仅在于将酶在4℃下放置2周后进行实验以比较蛋白质稳定性。泳道1是来自cas9
‑
hr3和grna hbb
‑
g1组合的消化模式。泳道2是来自cas9和grna hbb
‑
g1组合的消化模式。泳道3是来自cas9
‑
hr3和hbb
‑
g3组合的消化模式。泳道4是来自cas9和hbb
‑
g3组合的消化模式。泳道5是来自仅cas9
‑
hr的消化模式。泳道6是来自仅cas9的消化模式。泳道7是对照,其中既没有cas9也没有grna。
61.图19a
‑
g图示了通过cas9
‑
hr4、cas9
‑
hr8、仅cas9(nt)和没有cas9的对照(con)诱导h2bmneon融合物的基因组整合。图19a图示了h2b整合检测引物的设计。两组引物被设计用于在修复模板的5’和3’末端之外结合,与仅存在于基因组中而不存在于rt中的序列退火,而其他则与修复模板的特异性序列退火,并且不存在于未修饰的细胞中。图19b图示了琼脂糖凝胶,其显示了由5’引物扩增的pcr产物,表明内源性h2b用gfp成功标记。图19c图示了琼脂糖凝胶,其显示了由3’引物扩增的pcr产物,表明内源性h2b用gfp成功标记。图19d图示了由5’引物扩增的pcr产物的桑格测序序列迹线的吸光度。图分别按出现的顺序披露了seq id no 164
‑
165。图19e图示了由3’引物扩增的pcr产物的桑格测序序列迹线的吸光度。
图分别按出现的顺序披露了seq id no 166
‑
167。图19f图示了由5’引物扩增的pcr产物的测序比对,并按出现顺序分别公开了seq id no 155、154、153和160。图19g图示了由3’引物扩增的pcr产物的测序比对,并按出现顺序分别公开了seq id no 158、157、156和159。
62.图20图示了具有扩展功能的另外cas9
‑
hr融合蛋白的设计。
具体实施方式
63.包括关于crispr(成簇规律间隔短回文重复)/cas(crispr相关)系统的简要说明。首次在细菌和古细菌中发现的crispr/cas酶系统是针对病毒感染的免疫防御。在病毒感染期间,病毒dna片段被整合到crispr基因座中。这些整合的病毒dna片段被转录成指导rna(grna),它与病毒基因组在序列上互补。grna将cas酶引导至grna靶向的病毒基因组,其中cas蛋白切割病毒基因组,从而防御病毒感染。
64.crispr系统通常包含对靶dna序列具有特异性的grna和非特异性cas9蛋白。通常,grna包括两个不同的区段——crispr rna(crrna)和反式激活crispr rna(tracrrna)。crrna与靶dna序列互补,从而识别待切割的序列。并且tracrrna充当crrna
‑
cas9相互作用的支架。指导rna自然形成双链分子,其中crrna和tracrrna片段退火在一起。cas蛋白已被研究和工程构造为通过产生位点特异性双链断裂(dsb)进行基因编辑的工具。定制设计的grna引导cas蛋白在与grna序列互补的任何核酸基因座处生成dsb。cas蛋白已显示可以成功地在真核细胞中引入核苷酸变化、缺失、插入和置换。
65.使用crispr和cas9蛋白编辑核酸受到细胞内源性修复机制的限制。dsb优先由nhej修复。与nhej相关的修复位点处的意外插入和缺失使得基于遗传的疗法的发展不受欢迎。可替代地,如果产生的dsb被切除从而产生长(<200bp)3’突出端,则内源性修复途径将被迫使用hr。通过添加包含位于所需插入或缺失侧翼的同源臂的多核苷酸(模板序列),可以实现1
‑
1000bp的dna在任意位置的无错误靶向插入和缺失。
66.同源定向修复是无错误的,并且能够在给定的基因组中插入或缺失特定的dna序列。
67.此外,hdr降低了由crispr和cas9酶系统引入的dsb引起的细胞毒性。细胞毒性取决于p53肿瘤抑制途径,因为p53功能的抑制或丧失大大降低了人多能干细胞(hpsc)和永生化视网膜色素上皮(rpe)细胞中的细胞毒性。由于p53功能的永久性丧失对细胞有一些严重影响,包括基因组不稳定性、细胞稳态改变和体内癌症发病率增加,因此一种解决方案是通过小分子或显性阴性抑制剂的过表达对p53进行瞬时抑制。然而,体内p53的瞬时抑制具有挑战性,并可能产生不良副作用。因此,产生无毒的cas9酶对于体内应用是期望的。
68.本文公开了与在正常细胞和跟遗传疾病相关的细胞中选择性靶向和编辑内源性核酸区段相关的组合物和方法,其具有降低的细胞毒性。靶向的内源性核酸被切割、消化和通过hdr编辑。grna将包含cas蛋白部分和人exo1酶的蛋白质融合复合物引导至特定的内源性核酸区段,在那里所述蛋白质融合复合物引入切割和消化,在靶向的内源性核酸区段上留下3’或5’突出端。当细胞进一步被呈递多核苷酸片段时,突出端允许hdr速率增加,所述片段与靶向和消化的内源性核酸区段具有一定程度的序列同源性。
69.本文公开了组合物,其中靶向的内源性核酸位于已知的疾病基因座。靶向的已知疾病基因座被切割、消化和通过hdr编辑。grna将包含cas蛋白部分和人exo1酶的蛋白质融
合复合物引导至特定的已知疾病基因座,在那里所述蛋白质融合复合物引入切割和消化,在靶向的内源性核酸区段上留下3’或5’突出端。当细胞进一步被呈递多核苷酸片段时,突出端允许hdr速率增加,所述片段与靶向和消化的内源性核酸区段具有一定程度的序列同源性。融合蛋白组成
70.本文公开的组合物和方法的一些方面涉及至少一种经修饰的多肽,其包含与诸如人exo1外切核酸酶或其他外切核酸酶(例如mre11、exol、exoiii、exovii、exot、dna2、ctip、trex1、trex2、apollo、rece、recj、t5、lexo、recbcd和mungbean)的外切核酸酶的片段偶联的可编程内切核酸酶(例如cas9)或其他crispr相关可编程内切核酸酶,以相对于未修饰的可编程核酸内切酶(如crispr
‑
cas9系统中的cas9酶)的细胞毒性降低细胞毒性。cas9蛋白
71.多肽(融合蛋白)包含通过肽基接头共价连接的可编程内切核酸酶(例如cas9)、其他crispr相关可编程内切核酸酶、其他位点特异性内切核酸酶或其片段和外切核酸酶(例如人exo1外切核酸酶)或其片段。如本文所用,“cas9”、“cas9结构域”或“cas9片段”是指包含cas9蛋白或其片段(例如包含cas9的活性dna切割结构域的蛋白)的rna指导的核酸酶。cas9核酸酶有时被称为casn1核酸酶或crispr(成簇规律间隔短回文重复)相关核酸酶。cas9核酸酶序列和结构是本领域普通技术人员众所周知的。cas9直向同源物已在各种物种中得到描述,包括但不限于酿脓链球菌和嗜热链球菌。野生型(未修饰)cas9可以来自下表1中列出的任何序列。表1中列出的cas9蛋白质序列并不意味着是限制性的。其他合适的cas9核酸酶和蛋白质序列对本领域普通技术人员来说是显而易见的。表1.各种cas9的肽序列。
72.此外,在一些实施方案中,保留dna切割功能的cas9或其他可编程核酸酶的片段可用于产生融合蛋白。例如,cas9或其他可编程核酸酶多肽片段与野生型cas9至少约70%相同、至少约80%相同、至少约90%相同、至少约95%相同、至少约96%相同、至少约97%相同、至少约98%相同、至少约99%相同、至少约99.5%相同或至少约99.9%相同。在一些实施方案中,cas9片段与野生型cas9相比可具有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、21、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、
41、42、43、44、45、46、47、48、49、50或更多个氨基酸变化。
73.本文公开的cas9酶或其他可编程核酸酶还包含至少一个核定位信号(nls),其是一种氨基酸序列,附接于蛋白质以通过核转运输入到细胞核中。通常,nls包含暴露在蛋白质表面上的带正电荷的赖氨酸或精氨酸的一个或多个短序列。这些类型的经典nls可以进一步分类为单分或二分。两者之间的主要结构差异在于二分nls中的两个碱性氨基酸簇被相对较短的间隔序列隔开(因此二分
‑
2部分),而单分nls则不然。在一些实施方案中,nls包含sv40大t
‑
抗原(单分nls)的序列pkkkrkv(seq id no:19)。在其他实施方案中,核质蛋白的nls包含序列kr[paatkkagqa]kkkk(seq id no:20)。还有许多其他类型的非经典nls。本文公开的不同类型的nls并不意味着是限制性的,本领域普通技术人员能够选择nls以连接到cas9蛋白。在一些实施方案中,cas9蛋白包含n末端nls。在其他实施方案中,cas9蛋白包含c末端nls。在其他实施方案中,cas9蛋白包含n末端和c末端nls。
[0074]
在一些实施方案中,其他crispr相关可编程内切核酸酶通常包括crispr相关(cas)多肽或cas核酸酶,包括1类cas多肽、2类cas多肽、i型cas多肽、ii型cas多肽、iii型cas多肽、iv型cas多肽、v型cas多肽和vi型crispr相关(cas)多肽、crispr相关rna结合蛋白或其功能片段。此外,适用于本公开的cas多肽通常包括cpf1(或cas12a)、c2c1、c2c2(或cas13a)、cas13、cas13a、cas13b、c2c3、casl、caslb、cas2、cas3、cas4、cas5、cas5e(casd)、cas6、cas6e、cas6f、cas7、cas8a、cas8al、cas8a2、cas8b、cas8c、csnl、csxl2、cas10、cas10d、caslo、caslod、casf、casg、cash、csyl、csy2、csy3、csel(casa)、cse2(casb)、cse3(case)、cse4(casc)、cscl、csc2、csa5、csn2、csm2、csm3、csm4、csm5、csm6、cmrl、cmr3、cmr4、cmr5、cmr6、csbl、csb2、csb3、csxl7、csxl4、csxlo、csxl6、csax、csx3、csxl、csxl5、csfl、csf2、csf3、csf4和cul966;其任何衍生物;其任何变体;及其任何片段。
[0075]
此外,适用于本文公开的融合蛋白组合物的其他位点特异性内切核酸酶通常包括锌指核酸酶(zfn);转录激活因子样效应核酸酶(talen);大范围核酸酶;rna结合蛋白(rbp);重组酶;翻转酶;转座酶;argonaute(ago)蛋白(例如,原核生物argonaute(pago)、古菌argonaute(aago)和真核生物argonaute(eago));或其任何功能片段。hexo1蛋白
[0076]
可编程核酸酶通常与外切核酸酶结构域相连,以实现本文公开的结果。许多外切核酸酶/可编程外切核酸酶组合与本文的公开内容一致。关于外切核酸酶,适合用作本技术中融合蛋白的一部分的某些示例性外切核酸酶包括mre11、exol、exoiii、exovii、exot、dna2、ctip、trex1、trex2、apollo、rece、recj、t5、lexo、recbcd和mungbean。还考虑了另外的合适的外切核酸酶。在某些实施方案中,人exo1(hexo1)在本文中用作融合蛋白的一部分。全长hexo1大致可以分为两个区域:n末端的核酸酶区域(1
‑
392)(seq id no:1)mgiqgllqfi keasepihvr kykgqvvavd tycwlhkgai acaeklakgeptdryvgfcm kfvnmllshg ikpilvfdgc tlpskkever srrerrqanllkgkqllreg kvsearecft rsinithama hkvikaarsq gvdclvapyeadaqlaylnk agivqaiite dsdllafgck kvilkmdqfg ngleidqarlgmcrqlgdvf teekfrymci lsgcdylssl rgiglakack vlrlannpdivkvikkighy lkmnitvped yingfirann tflyqlvfdp ikrkliplnayeddvdpetl syagqyvdds ialqialgnk dintfeqidd ynpdtampah
srshswddkt cqksanvssi whrnysprpe sgtvsdapql ke),以及c末端mlh2/msh1相互作用区域(393
‑
846)。在一些实施方案中,hexo1(seq id no:1)的n末端核酸酶区域用于通过肽基接头与具有至少一个nls的cas9共价连接。在其他实施方案中,本文使用保留核酸酶功能的seq id no:1或其他外切核酸酶结构域的片段。例如,该片段与seq id no:1至少约70%相同,至少约80%相同,至少约90%相同,至少约95%相同,至少约96%相同,至少约97%相同,至少约98%相同、至少约99%相同、至少约99.5%相同或至少约99.9%相同。在一些实施方案中,该片段与seq id no:1或其他未截短或未突变的结构域相比可具有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、21、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50或更多个氨基酸变化。hexo1的n末端核酸酶区域是示例性的,并且本领域普通技术人员可以将另外合适的exo1或其他外切核酸酶序列用于本文公开的目的。
[0077]
在某些情况下,使用接头将外切核酸酶(例如hexo1肽)连接到可编程内切核酸酶(例如cas9肽)和至少一个nls。在一些实施方案中,接头是接头肽。接头肽不仅用于连接蛋白质部分,而且在某些情况下还提供许多其他功能,例如维持结构域间的协同相互作用或保持生物活性(gokhale rs,khosla c.role of linkers in communication between protein modules.curr opin chem biol.2000;4:22
–
27;ikebe m,kambara t,stafford wf,sata m,katayama e,ikebe r.a hinge at the central helix of the regulatory light chain of myosin is critical for phosphorylation
‑
dependent regulation of smooth muscle myosin motor activity.j biol chem.1998;273:17702
–
17707;以及chen xy,zaro j,和shen wc.fusion protein linkers:property,design and functionality.adv drug deliv rev 2014;65,1357
‑
1369并入本文)。接头肽可分为平均长度分别小于或高达4.5
±
0.7、9.1
±
2.4和21.0
±
7.6个残基或更大的小、中和大接头,尽管还考虑了由这三个范围定义的集合内的任何地方的示例。在一些实施方案中,接头肽包含5至200个氨基酸。在其他实施方案中,接头肽包含5至25个氨基酸。在某些实施方案中,接头肽选自fl2x(由seq id no:122(ggtctccttaaacctgtcttgt)编码)、sla2x(由seq id no:123(ggaggtggaggctctggtggaggcggatca)编码)、ap5x(由seq id no:124(gcagaggctgcagccgctaaggcc)编码)、fl1x(由seq id no:125(gcagaggctgcagccgctaaggaggcagctgccgctaaggcc)编码)、sla1x(由seq id no:126(gcacctgctccagcgcccgcaccagctccc)编码)及其任何组合。在一些实施方案中,接头肽是sla2x。同样,这些公开的接头肽并不意味着是限制性的。本领域普通技术人员能够选择合适的接头肽。
[0078]
本文公开的融合蛋白可以在翻译后直接融合在一起或从在共同开放阅读框中编码公开的融合蛋白的多核苷酸(融合核苷酸)翻译而来。在一些实施方案中,将编码hexo1或其n末端核酸酶区域的第一核酸序列连接至编码所选接头肽的第二核酸序列的一端。此外,第二核酸序列的另一端与编码具有至少一个nls的cas9酶的第三核酸序列连接。通常,去除第一、第二和第三核酸序列的终止密码子。在一些实施方案中,第一、第二和第三核酸序列经过密码子优化或工程改造以在靶细胞中更有效地转染或表达。类似地,在某些情况下,内
含子序列被去除。
[0079]
图20图示了通过接头(l1、l2和l3)连接的具有核酸酶、cas9和其他功能结构域的各种排列的示例性融合蛋白。融合蛋白的其他非限制性示例包括:hexo1
‑
cas9
‑
dn1s(或反向dn1s
‑
cas9
‑
hr);hexo1
‑
cas9
‑
dn1s
‑
孪蛋白(1
‑
110)(或dn1s
‑
cas9
‑
hr
‑
孪蛋白);hexo1
‑
cas9
‑
孪蛋白(1
‑
110)(或cas9
‑
hexo1
‑
孪蛋白);hexo1
‑
cas9
‑
pcv(或pcv
‑
cas9
‑
hexo1);hexo1
‑
cas9
‑
pcv
‑
孪蛋白(1
‑
110)(或pcv
‑
cas9
‑
hexo1
‑
孪蛋白);和hexo1
‑
cas9
‑
ctip(1
‑
296)(或ctip
‑
cas9
‑
hexo1)。
[0080]
在一些实施方案中,hexo1
‑
cas9
‑
dn1s(或反向dn1s
‑
cas9
‑
hr)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls(在图20中记为nls)的cas9随后通过接头2(tgs或其他)与在c末端添加nls序列的人p53(1231
‑
1644)的片段融合的融合物。在一些实施方案中,cas9
‑
hr和cas9
‑
dn1s可以在同源重组途径的不同步骤中起作用。在一些实施方案中,相对于cas9
‑
hr或cas9
‑
dn1s,hr
‑
cas9
‑
dn1s可具有增加的无错误编辑效率。在一些实施方案中,当与单独的cas9相比时,相对于用cas9
‑
dn1s观察到的增加,细胞毒性可以大大降低。
[0081]
在一些情况下,hexo1
‑
cas9
‑
dn1s
‑
孪蛋白(1
‑
110)(或dn1s
‑
cas9
‑
hr
‑
孪蛋白)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls的cas9随后通过接头2(tgs或其他)与人p53的片段(1231
‑
1644)融合的融合物。dn1s可以在其c末端添加nls,nls然后可以通过l3(任何序列)与孪蛋白(1
‑
110)融合,或者在其c末端与具有nls序列的孪蛋白融合,该nls序列可以通过l3融合到dn1s。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
dn1s
‑
孪蛋白(1
‑
110)(或dn1s
‑
cas9
‑
hr
‑
孪蛋白)的细胞毒性可以降低。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
dn1s
‑
孪蛋白(1
‑
110)(或dn1s
‑
cas9
‑
hr
‑
孪蛋白)的无错误编辑效率可以提高。在一些实施方案中,由于hexo1
‑
cas9
‑
dn1s
‑
孪蛋白的孪蛋白的翻译后调节将核酸酶活性限制在s/g2期(此时细胞中内源性hr最高),与cas9相比,hexo1
‑
cas9
‑
dn1s
‑
孪蛋白(1
‑
110)(或dn1s
‑
cas9
‑
hr
‑
孪蛋白)的无错误编辑效率可以提高。
[0082]
在一些实施方案中,hexo1
‑
cas9
‑
孪蛋白(1
‑
110)(或cas9
‑
hexo1
‑
孪蛋白)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls、具有或缺乏c末端nls序列的cas9随后通过接头2(tgs或其他)与具有或缺乏c末端nls序列的孪蛋白(1
‑
110)片段融合的融合物。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
孪蛋白(1
‑
110)(或cas9
‑
hexo1
‑
孪蛋白)包括降低的细胞毒性和提高的无错误编辑效率。
[0083]
在一些实施方案中,hexo1
‑
cas9
‑
pcv(或pcv
‑
cas9
‑
hexo1)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls、具有或缺乏c末端nls序列的cas9随后通过接头2(tgs或其他)与pcv融合的融合物。在一些实施方案中,pcv可以结合特定的ssdna序列,从而将修复模板连接到cas9复合物。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
pcv包括提高的无错误编辑效率。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
pcv包括降低的细胞毒性。
[0084]
在一些实施方案中,hexo1
‑
cas9
‑
pcv
‑
孪蛋白(1
‑
110)(或pcv
‑
cas9
‑
hexo1
‑
孪蛋白)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls、具有或缺乏c末端nls序列的cas9随后通过接头2(tgs或其他)与pcv(其然后可以与孪蛋白(1
‑
110)的片段融合)融合的融合物。在一些实施方案中,与cas9相比,hexo1
‑
cas9
‑
pcv
‑
孪蛋白
(1
‑
110)(或pcv
‑
cas9
‑
hexo1
‑
孪蛋白)包括更高的无错误编辑效率。在一些实施方案中,由于核酸酶活性限制于s/g2期,因此hexo1
‑
cas9
‑
pcv
‑
孪蛋白(1
‑
110)(或pcv
‑
cas9
‑
hexo1
‑
孪蛋白)包含比cas9更高的无错误编辑效率。
[0085]
在一些实施方案中,hexo1
‑
cas9
‑
ctip(1
‑
296)(或ctip
‑
cas9
‑
hexo1)可以是hexo1(1
‑
352)通过接头1(fl1x、ap5x或其他)与具有或缺乏n末端flag+nls、具有或缺乏c末端nls序列的cas9随后通过接头2(tgs或其他)与ctip融合的融合物。在一些实施方案中,与没有ctip的cas9相比,ctip可以提高无错误编辑效率。在一些实施方案中,与cas9相比,ctip可以通过在被阻断的dsb(双链断裂)下游结合并使用3
’‑5’
外切核酸酶活性针对断裂向后切除而提高无错误编辑效率。
[0086]
exoi的大肠杆菌(e.coli)版本
[0087]
在某些实施方案中,exoi的大肠杆菌(e.coli)版本(e.coli exoi)在本文中用作融合蛋白的一部分。与hexo1的5’到3’外切核酸酶活性相反,大肠杆菌exo1具有3’到5’外切核酸酶活性。大肠杆菌exoi cas9融合可以产生比传统cas9长得多的缺失。核酸序列
[0088]
与本公开一致的一些核苷酸构建体包含编码外切核酸酶例如hexo1的核酸。此外,与本公开一致的一些核苷酸构建体包含编码可编程内切核酸酶例如cas9或其他crispr相关的可编程内切核酸酶的核酸。在一些实施方案中,编码hexo1或其n末端核酸酶区域的核酸序列是非天然存在的,但由其编码的hexo1或其n末端核酸酶区域具有天然存在的氨基酸序列。在一些情况下,核酸序列不同于天然存在的hexo1或其n末端核酸酶区域核酸序列,但由于密码子简并性编码与hexo1或其n末端核酸酶区域相同的多肽。类似地,编码具有至少一个nls的cas9酶的第三核酸序列是非天然存在的,但由其编码的cas9蛋白具有天然存在的氨基酸序列。在一些情况下,核酸序列不同于天然存在的cas9核酸序列,但由于密码子简并性编码与cas9相同的多肽。核糖核蛋白(rnp)
[0089]
核糖核蛋白(rnp)通常包括至少两个部分:一个部分包含可编程内切核酸酶,例如cas9,或其他crispr相关的可编程内切核酸酶;另一个部分包含grna或其他特异性传递的核酸。通常,野生型cas9酶或其他cas或非cas可编程内切核酸酶可以是crispr
‑
cas9系统的一个部分。通过接头肽与hexo1片段偶联的经修饰cas9蛋白也可以是crispr
‑
cas9系统的一个部分。此外,经修饰的cas9蛋白和grna可以形成核糖核蛋白(rnp)。grna
[0090]
本文使用的核糖核酸包含用于将核糖核酸引导至基因上的靶位点的序列和用于结合内切核酸酶例如cas9酶的另一序列。通常,核糖核酸是grna。在一些实施方案中,grna是合成grna(sgrna)。grna将融合蛋白复合物引导至dna分子的靶向核苷酸序列。grna是一种短合成rna,由cas结合所需的支架序列和用户定义的约20个核苷酸间隔区组成,该间隔区定义了要修饰的基因组靶。在某些实施方案中,可以设计grna的间隔区以识别hbb基因的外显子1。因此,可以通过简单地改变grna中存在的靶序列来改变cas蛋白的基因组靶。
[0091]
有几种方法可以将grna递送到细胞中。一种是将grna作为质粒dna递送到细胞中。在一些实施方案中,可以将编码融合蛋白的核酸克隆到一个具有编码靶向感兴趣基因的经设计的grna的核酸序列的质粒或其他合适的载体中。
[0092]
下面提供了代表性grna成分的列表。表2.grna序列列表。
hdr模板序列
[0093]
基因组稳定性需要正确有效地修复dsb。在真核细胞中,dsb的机械修复主要通过两种途径发生:非同源末端连接(nhej)和同源定向修复(hdr)。nhej是典型的不依赖于同源性的途径,因为它最多只涉及一到几个互补碱基的比对以重新连接两端,而hdr使用更长段的序列同源性来修复dna损伤。由于dna的受损和完整供体链之间需要更高的序列同源性,因此hdr是更准确的dsb修复机制。如果用于修复的dna模板与dsb处的原始dna序列相同,则该过程是无错误的,或者它可以将非常特定的突变引入受损的dna。
[0094]
如上所述,hdr方法在基因组工程中提供了极大的自由度,允许少至单碱基突变,多至插入或缺失千碱基(kb)的dna。在真核生物中,hdr速率受两种不同途径之间的竞争控制:同源重组(hr)和非同源末端连接(nhej)。这两种途径之间的竞争始于mrn/ctip复合物或ku 70/80异二聚体的竞争性结合。如果mrn/ctip首先结合,则它们会招募其他蛋白质,包括外切核酸酶i(exoi),它们具有5
’‑
>3’外切核酸酶活性20。在断裂的每一侧通过exo1或dna2对双链dna断裂进行5’末端切除,使dsb由hr途径修复。可替代地,如果ku 70/80异二聚体结合,则它可以招募其他nhej途径成员,包括dna连接酶iv,并最终通过nhej修复双链断裂。
[0095]
将crispr
‑
cas9系统递送至细胞时,需要将hdr模板序列递送至细胞中。用于生成特定突变或将新元件插入基因的hdr模板需要在将被修饰的靶序列周围具有一定量的同源性。在一些实施方案中,5’和3’同源臂始于crispr诱导的dsb。一般来说,修饰的插入位点可以非常靠近dsb,如果可能的话,理想情况下小于10bp。在一些实施方案中,hdr模板序列的5’和3’同源臂与靶序列至少80%相同。此外,在一些实施方案中,单链供体寡核苷酸(ssdon)用于较小的插入。ssdon的每个同源臂可以包含约30
‑
80bp的核苷酸序列。同源臂的长度不是限制性的,本领域普通技术人员可以根据感兴趣的基因座和实验系统调整长度。
对于较大的插入,如荧光蛋白或选择盒,双链供体寡核苷酸(dsdon)可用作hdr模板序列。在一些实施方案中,ssdon的每个同源臂可包含约800
‑
1500bp核苷酸序列。为了防止cas9酶切割hdr模板,在一些实施方案中,可以在hdr模板的前间区序列邻近基序(pam)序列中引入单碱基突变。递送方法
[0096]
使用几种不同的方法将核糖核蛋白和ssdon或其他核酸递送到切割位点,例如转染。转染方法可用于将crispr
‑
cas9或其他可编程内切核酸酶组分递送至细胞。一些示例性方法可用于将所公开的经修饰的crispr
‑
cas9系统递送至细胞,并且与本领域普通技术人员已知的本公开内容一致的另外方法可以根据细胞的类型和crispr
‑
cas9组分的形式选择特定的方法。
[0097]
递送可以分为两大类:货物(cargo)和递送媒介。关于crispr/cas9货物,通常可用三种方法:(1)编码cas9蛋白或其他可编程内切核酸酶和指导rna的dna质粒,(2)用于cas9或其他可编程内切核酸酶翻译的mrna,以及单独的指导rna,以及(3)具有指导rna的cas9蛋白质或其他可编程内切核酸酶(核糖核蛋白复合物)。所使用的递送媒介通常会决定可以包装这三种货物中的哪一种,以及该系统是否可用于体外和/或体内。
[0098]
用于递送基因编辑系统货物的媒介可分为三大类:物理递送、病毒载体和非病毒载体。最常见的物理递送方法是显微注射、电穿孔和核转染。电穿孔能够在使用基于脂质的传递系统难以转化的细胞类型中递送crispr机器。对细胞施加受控的短电脉冲在细胞膜上形成孔,允许外来物质进入。核转染是电穿孔的一种变体,其中优化了电脉冲,使得细胞的核膜也形成孔。因此,crispr组分直接递送到细胞核内。显微注射通常用于将cas9或其他可编程内切核酸酶和grna核糖核蛋白复合物注射到胚胎中,尽管它也可用于细胞中。斑马鱼、小鼠和最近的人胚胎已使用这种技术进行了操作。
[0099]
病毒递送载体包括专门工程改造的腺伴随病毒(aav),以及全尺寸的腺病毒和慢病毒载体。特别是在体内工作中,病毒载体受到青睐,是最常见的crispr/cas9递送载体。依赖病毒属和细小病毒科的aav是单链dna病毒,已广泛用于基因治疗。虽然lv和adv明显不同,但它们用于递送crispr/cas9组分的方式十分相似。在lv递送的情况下,主干病毒是hiv的前病毒;对于adv递送,主干病毒是已知adv的许多不同血清型之一。lv和adv都可以感染分裂细胞和非分裂细胞;然而,与lv不同的是,adv不会整合到基因组中。这在基于crispr/cas9的编辑中对于限制脱靶效应是有利的。与aav颗粒的情况一样,lv和adv均可用于体外、离体和体内应用,从而简化了功效和安全性测试。在机制上,这类crispr/cas9递送类似于上述的aav递送。含有所期望的cas9和sgrna的完整病毒颗粒是通过转化hek293 t细胞产生的。然后使用这些病毒颗粒感染靶细胞类型。lv/adv递送和aav递送的最大区别在于颗粒的大小;lv和adv的直径大约为80
‑
100nm。与aav的20nm直径相比,这些系统对更大的插入有更好的耐受性。在考虑crispr/cas9时,用于不同大小的cas9构建体或用于多重基因组编辑的多个sgrna的额外包装空间比aav递送系统具有显著优势。
[0100]
病毒载体可以是经修饰的病毒载体,可替代地,它可以是未修饰的载体。通常,经修饰的病毒载体是经遗传修饰的载体。经修饰的病毒载体可显示免疫原性降低、载体在血流中的持久性增加或巨噬细胞和抗原呈递细胞对载体的摄取受损。
[0101]
经修饰的病毒载体还可包含聚合物、脂质、肽、磁性纳米颗粒(mnp)、另外的化合物
或其组合。聚合物、脂质或磁性纳米颗粒可以附着到病毒载体的衣壳上。聚合物可以是聚乙二醇(peg)。聚合物可以是n
‑
[2
‑
羟基丙基]甲基丙烯酰胺(hpma)、聚(2
‑
(二甲基氨基)乙基甲基丙烯酸酯)(pdmaema)或精氨酸接枝的生物可还原聚合物(abp)。肽可以是细胞穿透肽、细胞粘附肽或与细胞上的受体结合的肽。细胞可以是肿瘤细胞。可以使用任何合适的细胞穿透肽。细胞穿透肽的实例包括但不限于聚赖氨酸肽和聚精氨酸肽。细胞粘附肽可以是精氨酰甘氨酰天冬氨酸(rgd)肽。另外的化合物可以是与细胞上的受体结合的化合物,例如叶酸。
[0102]
在一些情况下,经修饰的病毒载体是经遗传修饰的载体。经遗传修饰的载体可具有降低的免疫原性、降低的基因毒性、增加的负载能力、增加的转基因表达或其组合。在一些情况下,经遗传修饰的病毒载体是假型病毒载体。假型病毒载体可具有至少一种外源病毒包膜蛋白。外源病毒包膜蛋白可以是来自狂犬病病毒属、沙粒病毒属、嗜肝性dna病毒、黄病毒属、副粘病毒属、杆状病毒属、丝状病毒或甲病毒属的包膜蛋白。外源病毒包膜蛋白可以是水疱性口炎病毒(vsv)的糖蛋白g。在一些情况下,外源病毒包膜蛋白是经遗传修饰的病毒包膜蛋白。经遗传修饰的病毒包膜蛋白可以是非天然存在的病毒包膜蛋白。
[0103]
在一些实施方案中,病毒载体是病毒样颗粒(vlp)。vlp类似于病毒,但不具有传染性,因为它们不包含病毒遗传物质。vlp是由多种病毒家族的组分产生的,所述病毒家族包括细小病毒科(例如腺伴随病毒),逆转录病毒科(例如hiv),黄病毒科(例如丙型肝炎病毒)和噬菌体。vlp可以在多种细胞培养系统中生产,所述细胞培养系统包括细菌、哺乳动物细胞系、昆虫细胞系、酵母和植物细胞。
[0104]
对于非病毒载体递送载体,本文可使用脂质纳米颗粒/脂质体。脂质可以是阳离子脂质、阴离子脂质或中性脂质。脂质可以是脂质体、小单层脂囊(suv)、脂质包膜、类脂质或脂质纳米颗粒(lnp)。脂质可以与核酸混合以形成脂质复合物(核酸
‑
脂质体复合物)。脂质可以与核酸缀合。脂质可以是非ph敏感脂质或ph敏感脂质。脂质还可包含聚乙二醇(peg)。
[0105]
阳离子脂质可以是单价阳离子脂质,例如n
‑
[1
‑
(2,3
‑
二油酰基氧基)丙基]
‑
n,n,n
‑
三甲基氯化铵(dotma)、[1,2
‑
双(油酰基氧基)
‑3‑
(三甲基氨基)丙烷](dotap)或3β[n
‑
(n',n'
‑
二甲氨基乙烷)
‑
氨基甲酰基]胆固醇(dc
‑
chol)。阳离子脂质可以是多价阳离子脂质,例如二
‑
十八烷基
‑
酰胺基
‑
甘氨酰基
‑
精胺(dogs)或{2,3
‑
二油酰基氧基
‑
n
‑
[2(精胺甲酰胺)乙基]
‑
n,n
‑
二甲基
‑
l
‑
三氟乙酸丙铵}(dospa)。
[0106]
阴离子脂质可以是磷脂或二油酰基磷脂酰甘油(dopg)。磷脂的实例包括但不限于磷脂酸、磷脂酰甘油或磷脂酰丝氨酸。在一些情况下,阴离子脂质还包含二价阳离子,例如ca2+、mg2+、mn2+和ba2+。
[0107]
阳离子脂质或阴离子脂质还可包含中性脂质。中性脂质可以是二油酰基磷脂酰乙醇胺(dope)或二油酰基磷脂酰胆碱(dopc)。在一些情况下,辅助脂质与带电脂质的组合使用产生更高的转染效率。
[0108]
脂质体还可包含聚合物、脂质、肽、磁性纳米颗粒(mnp)、另外的化合物或其组合。聚合物、脂质或磁性纳米颗粒可以附着到脂质体或整合到脂质体膜中。聚合物可以是聚乙二醇(peg)。聚合物可以是n
‑
[2
‑
羟基丙基]甲基丙烯酰胺(hpma)、聚(2
‑
(二甲基氨基)乙基甲基丙烯酸酯)(pdmaema)或精氨酸接枝的生物可还原聚合物(abp)。肽可以是细胞穿透肽、细胞粘附肽或与细胞上的受体结合的肽。细胞可以是肿瘤细胞。可以使用任何合适的细胞
穿透肽。细胞穿透肽的实例包括但不限于聚赖氨酸肽和聚精氨酸肽。细胞粘附肽可以是精氨酰甘氨酰天冬氨酸(rgd)肽。另外的化合物可以是与细胞上的受体结合的化合物,例如叶酸。试剂盒
[0109]
本文公开了与本文所述的一种或多种方法和组合物一起使用的试剂盒和制品。试剂盒可以包含在相容的药用赋形剂中配制并放置在合适的容器中的本文所述的多核苷酸组合物。
[0110]
试剂盒可包括载器、包装或容器,其被隔开以接收一个或多个容器,例如小瓶、管等,所述一个或多个容器中的每个包含将在本文所述方法中使用的单独元件之一。合适的容器包括例如瓶子、小瓶、注射器和试管。容器可以由多种材料制成,例如玻璃或塑料。
[0111]
试剂盒可包括鉴别说明、标签或包装插页。标签或包装插页可列出试剂盒的内容物或免疫组合物、与其在本文所述的方法中的使用相关的说明或其组合。标签可以位于容器上或与容器相关联。当形成标签的字母、数字或其他字符被附着、模制或蚀刻到容器本身中时,标签可以位于容器上。当标签存在于也保持容器的受器或载器中时,例如作为包装插页,标签可以与容器相关联。在某些情况下,标签用于表明内容物将用于特定的治疗应用。
[0112]
本文的试剂盒还可包含一种或多种用于将多核苷酸序列递送至细胞、组织或器官的试剂。应用
[0113]
可以使用本文公开的递送方法之一将公开的rnp引入细胞中以诱导细胞中dna的同源重组。此外,可以使用本文公开的递送方法之一将公开的rnp引入细胞中以在体外或离体细胞中诱导hdr。dna分子与rnp接触。由grna指导的经修饰的cas9蛋白通过在由grna与dna分子杂交确定的位置处切割来引入dsb。hexo1肽部分消化切割的dna分子,留下3’或5’突出端。hdr模板序列包含一定程度的序列同源性,因为消化的dna分子促进hdr并用作hdr的模板。在hdr之后,细胞中的dna分子在发生同源重组的区域包含与hdr模板相同的序列。
[0114]
通过在细胞中诱导hdr,野生型cas9蛋白和grna引起的细胞毒性降低。细胞毒性可以通过几种细胞活力测定来测量。在一些实施方案中,使用四唑还原测定。多种四唑化合物已被用于检测活细胞。最常用的化合物包括:mtt、mts、xtt和wst
‑
1。这些化合物分为两个基本类别:1)带正电且容易渗透活的真核细胞的mtt和2)带负电且不易渗透细胞的诸如mts、xtt和wst
‑
1之类的化合物。后一类(mts、xtt、wst
‑
1)通常与中间电子受体一起使用,该受体可以从细胞质或质膜转移电子,以促进四唑还原成有色甲臜产物。例如,代谢活跃的活细胞将mtt转化为紫色甲臜产物,在570nm附近具有最大吸光度。当细胞死亡时,它们会失去将mtt转化为甲臜的能力,因此颜色的形成可以作为仅活细胞的有用且方便的标记。
[0115]
在其他实施方案中,使用刃天青还原测定。刃天青是一种可渗透细胞的氧化还原指示剂,可用于通过类似于使用四唑化合物的方案来监测活细胞数量。刃天青可以溶解在生理缓冲液中(产生深蓝色溶液),并以均质形式直接添加到培养细胞中。代谢活跃的活细胞可将刃天青还原成呈粉红色荧光的试卤灵产物。产生的试卤灵的数量与活细胞的数量成正比,可以使用配备530nm或560nm激发/590nm发射滤光片组的微孔板荧光计进行量化。波长可根据不同类型的细胞和实验设计进行调整。试卤灵也可以通过测量吸光度的变化来量化;然而,吸光度检测并不常用,因为它远不如测量荧光灵敏。
[0116]
此外,本文公开的rnp用于治疗病因被追踪到染色体异常基因座的疾病。在某些实施方案中,生物样品获自患有疾病的受试者。从生物样品中提取dna并对其测序以确定染色体异常的基因座。从受试者中分离出含有染色体异常的原代细胞并进行离体培养。使用本文公开的递送方法之一将rnp递送到所述培养的原代细胞中。hdr模板序列也被递送到培养的原代细胞中。在一些实施方案中,grna部分包含至少10个与染色体异常的靶基因座互补的核苷酸。hdr模板序列包含侧接有5’同源区和3’同源区的整合盒,其中5’同源区和3’同源区与靶基因座的相邻区段表现出至少80%的同一性。hdr模板的整合盒包含野生型序列,该序列对应于在原代细胞中检测到的染色体异常基因座。在递送rnp后,grna将蛋白质融合复合物引导至靶基因座,其中经修饰的cas蛋白部分通过切割grna识别的所述靶基因座来创建dsb。核酸酶部分部分消化染色体异常的切割基因座,留下3’突出端。hdr模板序列的存在通过hdr促进内源性修复。筛选并选择具有取代染色体异常的野生型序列的原代细胞以重新引入受试者。
[0117]
在一些实施方案中,原代细胞选自包括以下的组:t细胞、b细胞、树突细胞、自然杀伤细胞、自然杀伤细胞、巨噬细胞、中性粒细胞、嗜酸性粒细胞、嗜碱性粒细胞、肥大细胞、造血祖细胞、造血干细胞(hsc)、红细胞、血干细胞、内胚层干细胞、内胚层祖细胞、内胚层前体细胞、分化的内胚层细胞、间充质干细胞(msc)、间充质祖细胞、间充质前体细胞、分化的间充质细胞、肝细胞祖细胞、胰腺祖细胞、肺祖细胞、气管祖细胞、骨细胞、软骨细胞、肌肉细胞、脂肪细胞、基质细胞、成纤维细胞和真皮细胞。
[0118]
此外,在一些实施方案中,grna被配置为识别人hbb基因的外显子1。hdr模板被配置为与功能性人hbb基因具有5’和3’臂同源性。在其他实施方案中,grna被配置为识别cftr的区域并且hdr模板被设计为与功能性cftr基因具有5’和3’臂同源性。
[0119]
请参阅表3中分别列出基因的突变基因座的单基因遗传障碍列表。人单基因疾病、遗传模式和相关基因的示例。表3.
[0120]
此外,本文公开的rnp用于引入遗传修饰以赋予针对疾病的免疫力。从受试者获得生物样品。提取dna并对靶向遗传修饰的基因座进行测序。受试原代细胞被离体分离和培养。rnp和hdr模板序列被递送到所述培养的原代细胞中。grna部分将rnp引导至靶基因座以启动dsb的形成和dna消化以产生3’突出端。hdr模板包含侧接有5’同源区和3’同源区的整合盒,其中5’同源区和3’同源区与靶基因座的相邻区段表现出至少80%的同一性。整合盒包含野生型序列,该序列不同于靶基因座处的受试者的序列。多核苷酸的存在通过hdr促进内源性修复。筛选并选择含有由多核苷酸编码的野生型序列的原代细胞以重新引入受试者。某些定义
[0121]
除非上下文另外明确规定,否则如本文和所附权利要求书中所使用,单数形式“一个/一种(a/an)”和“所述”包括复数指示物。还应注意,权利要求可以被修改成排除任何任选要素。因此,此陈述旨在作为使用与权利要求要素的叙述有关的排他性术语如“单独”、“仅”等或使用“否定型”限定的前提基础。
[0122]
如本文所用,术语“多肽”、“肽”和“蛋白质”在本文中关于氨基酸残基的聚合物通常可互换使用。蛋白质通常是指从编码开放阅读框翻译出来的全长多肽,或加工成其成熟形式的全长多肽,而多肽或肽非正式地指蛋白质的降解片段或加工片段,尽管如此其仍然唯一或可识别地映射到特定蛋白质。多肽可以是通过相邻氨基酸残基的羧基基团和氨基基
团之间的肽键结合在一起的氨基酸的单一线性聚合物链。例如,可以通过添加碳水化合物、磷酸化等来修饰多肽。蛋白质可以包含一种或多种多肽。
[0123]
如本文所用,术语“片段”、“结构域”或等同术语是指蛋白质的一部分,其长度小于蛋白质的全长并保持蛋白质的功能。此外,当蛋白质的一部分再次与蛋白质进行局部序列排比检索(blast)时,蛋白质序列的该部分将与蛋白质序列的一部分以至少80%的同一性比对。
[0124]
如本文所用,术语“多核苷酸”、“核酸”、“寡核苷酸”或等同术语是指包含核苷酸碱基单体的聚合排列的分子,其中单体序列定义了多核苷酸。多核苷酸可包括产生脱氧核糖核酸(dna)的脱氧核糖核苷酸聚合物和产生核糖核酸(rna)的核糖核苷酸聚合物。多核苷酸可以是单链或双链的。当单链时,多核苷酸可对应于基因的有义或反义链。单链多核苷酸可以与靶多核苷酸的互补部分杂交以形成双链体,该双链体可以是同源双链体或异源双链体。多核苷酸的长度在任何方面都没有限制。核苷酸之间的连接可以是核苷酸间型磷酸二酯连接,或任何其他类型的连接。多核苷酸可以通过生物学方法(例如,酶促)在体内(在细胞中)或体外(在无细胞系统中)产生。可以使用无酶系统化学合成多核苷酸。多核苷酸可以是酶促可延伸的或酶促不可延伸的。
[0125]
如本文所用,术语“载体”、“媒介”、“构建体”和“质粒”用于指可繁殖并用于将一个或多个核酸区段从一种生物体转移至另一种生物体的任何重组多核苷酸分子。载体通常包含介导载体繁殖和操作的部分(例如,一个或多个复制起点、赋予药物或抗生素抗性的基因、多克隆位点、可操作连接的启动子/增强子元件(其能够表达克隆的基因)等)。载体通常是重组核酸分子,通常来源于噬菌体或植物或动物病毒。质粒和粘粒是指两种这样的重组载体。“克隆载体”或“穿梭载体”或“亚克隆载体”包含促进亚克隆步骤的可操作地连接的部分(例如,包含多个限制性内切核酸酶靶序列的多克隆位点)。取决于载体类型或应用类型,核酸载体可以是线性分子或环状分子。一些环状核酸载体可以在递送到细胞中之前有意地线性化。
[0126]
如本文所用,术语“基因”通常是指多核苷酸元件的组合,当以天然或重组方式可操作地连接时,所述元件提供一些产物或功能。术语“基因”应作广义解释,可包括基因的mrna、cdna、crna和基因组dna形式。在一些用途中,术语“基因”涵盖转录序列,包括5’和3’非翻译区(5
’‑
utr和3
’‑
utr)、外显子和内含子。在一些基因中,转录区将包含编码多肽的“开放阅读框”。在该术语的一些使用中,“基因”仅包含编码多肽所必需的编码序列(例如,“开放阅读框”或“编码区”)。在一些方面,基因不编码多肽,例如核糖体rna基因(rrna)和转移rna(trna)基因。在一些方面,术语“基因”不仅包括转录序列,而且还包括非转录区,包括上游和下游调节区、增强子和启动子。术语“基因”包括基因的mrna、cdna和基因组形式。
[0127]
如本文所用,术语“受试者”、“个体”或“患者”在本文中经常互换使用。“受试者”可以是包含表达的遗传物质的生物实体。生物实体可以是植物、动物或微生物,包括例如细菌、病毒、真菌和原生动物。受试者可以是体内获得或体外培养的生物实体的组织、细胞及其后代。受试者可以是哺乳动物。哺乳动物可以是人。可以诊断受试者或怀疑受试者具有疾病高风险。疾病可以是癌症。在一些情况下,不一定诊断受试者或怀疑受试者具有疾病高风险。
[0128]
如本文所用,术语“体内”用于描述在受试者体内发生的事件。
[0129]
如本文所用,术语“离体”用于描述在受试者体外发生的事件。不对受试者进行“离体”测定。相反,它是在从受试者分离的样品上进行的。对样品进行“离体”测定的示例是“体外”测定。
[0130]
如本文所用,术语“体外”用于描述在容器中发生的事件,所述容器容纳实验室试剂使得它与从中获得材料的活的生物源有机体分离。体外测定可以包括基于细胞的测定,其中使用活细胞或死细胞。体外测定还可以包括不使用完整细胞的无细胞测定。
[0131]“进行治疗(treating)”或“治疗(treatment)”是指治疗性治疗和预防性措施,其中目的是预防或减缓(减轻)目标病理病况或病症。需要治疗的人包括那些已经患有该病症的人,以及那些易于患有该病症的人,或者那些需要预防该病症的人。治疗益处可以指根除或改善症状或正在治疗的潜在病症。此外,通过消除或改善与潜在病症相关的一种或多种生理症状,可以获得治疗益处,从而在受试者中观察到了改善,尽管受试者仍然患有所述潜在病症。预防作用包括延迟、预防或消除疾病或病况的出现,延迟或消除疾病或病况的症状发作,减缓、停止或逆转疾病或病况的进展,或其任何组合。对于预防益处,处于发展特定疾病的风险中的受试者或报告一种或多种疾病生理症状的受试者可以接受治疗,即使可能尚未做出该疾病的诊断。
[0132]
本文以在数值之前带有术语“约”提供某些范围。术语“约”在本文中用于为其后的确切数字以及与所述术语后的数字接近或近似的数值提供文字支持,例如在它后面的数值的10%以内的数字。在确定数字是否接近或近似于具体列举的数字时,接近或近似的未列举数字可以是在所述数字出现的上下文中提供具体列举的数字的大致相等的数字。在提供数值范围的情况下,应当理解,除非上下文另有明确规定,否则所述范围的上限和下限之间的每个中间值(到下限单位的十分之一)以及所述范围内的任何其他陈述值或中间值包含在本文所述的方法和组合物内。这些较小范围的上限和下限可以独立地包括在较小范围内,并且也包括在本文所述的方法和组合物内,受到所述范围内的任何明确排除性限制。当所述范围包括所述极限的一个或两个时,排除那些所包括的极限中的一个或两个的范围也包括在本文所述的方法和组合物内。
[0133]
在整个公开内容中经常提到“cas9”。应理解,虽然cas9是特定实施方案,但也考虑了另外的可编程内切核酸酶,例如cas12或其他。因此,不应总是将提及cas9视为排除替代物或其他可编程内切核酸酶。
[0134]
类似地,经常提到“hexo1”。应当理解,虽然hexo1是一个特定的实施方案,但也考虑了另外的可编程内切核酸酶。因此,不应总是将提及hexo1视为排除替代物或其他外切核酸酶。
[0135]
除非另外定义,否则本文使用的所有技术和科学术语具有与本文所述的方法和组合物所属领域的普通技术人员通常理解的含义相同的含义。尽管与本文所述的那些方法和材料相似或等效的任何方法和材料也可用于实践或测试本文所述的方法和组合物中,但现在描述代表性的说明性方法和材料。
附图说明
[0136]
图1显示了9个cas9
‑
hr融合蛋白。第一融合蛋白包含通过接头fl2x与具有n末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第二融合蛋白包
含通过接头sla2x与具有n末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第三融合蛋白包含通过接头ap5x与具有n末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第四融合蛋白包含通过接头fl1x与具有n末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第五融合蛋白包含通过接头sla1x与具有n末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第六融合蛋白包含通过接头fl2x与具有n末端nls和c末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第七融合蛋白包含通过接头sla2x与具有n末端nls和c末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。以及第八融合蛋白包含通过接头ap5x与具有n末端nls和c末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。第九融合蛋白包含直接与具有n末端nls和c末端nls的cas9(seq id no:2
‑
18中的任何一个)偶联的hexo1(seq id no:1)。
[0137]
图2显示了用于核苷酸切割和替换的预期靶位点的实施方案。预期的靶位点距离人h2b基因6号染色体的3’末端约1kb。该图还显示了hdr模板的实施方案,它包含在5’末端与cmv启动子偶联并在3’末端与sv40 poly(a)偶联的嘌呤霉素抗生素抗性盒。此外,hdr模板包含上述预期靶位点的5’和3’同源区域。在pam序列中引入单个g
‑
>c突变以防止rnp切割hdr模板。
[0138]
图3显示了实验设计的实施方案。在96孔板中培养细胞,每孔接种约2.5x104个细胞。96孔板的每一列分别接受不同质粒的处理。因为每列包含8个孔,每个处理有8个重复。第一列中的细胞用编码如图1所示的第一融合蛋白的质粒转染;第二列中的细胞用编码如图1所示的第二融合蛋白的质粒转染;第三列中的细胞用编码如图1所示的第三融合蛋白的质粒转染;第四列中的细胞用编码如图1所示的第四融合蛋白的质粒转染;第五列中的细胞用编码如图1所示的第五融合蛋白的质粒转染;第六列中的细胞用编码如图1所示的第六融合蛋白的质粒转染;第七列中的细胞用编码如图1所示的第七融合蛋白的质粒转染;以及第八列中的细胞用编码如图1所示的第八融合蛋白的质粒转染。此外,第九列中的细胞用编码未修饰的cas9酶的质粒转染。第十列中的细胞用编码gfp的质粒转染。第十一列中的细胞是未经任何处理的对照。
[0139]
图4显示柱形图,其显示在嘌呤霉素选择之前测得的试卤灵荧光的归一化倍数变化。y轴显示归一化倍数变化数,x轴显示分别用编码8种融合蛋白的质粒、未修饰的cas9、gfp的质粒和未转染的对照进行的处理。由于最小的细胞毒性,预计来自对照处理的细胞具有最大的试卤灵荧光。将对照处理的试卤灵荧光测量值归一化为1,因此,它们的归一化倍数变化数为1。将每隔一个处理的试卤灵荧光测量值与对照处理进行比较,以获得归一化倍数变化数。还预期所有处理都具有一定程度的细胞毒性,因此,每个处理可以具有小于1的归一化倍数变化数。例如,与对照处理相比,野生型cas9处理显示出最小的倍数变化数,这意味着野生型cas9转染的细胞具有最少的试卤灵荧光。相比之下,用编码第七融合蛋白和gfp的质粒处理具有相似且最大的倍数变化数,这表明转染的细胞具有第二大的试卤灵荧光量。
[0140]
图5显示柱形图,其显示在用编码野生型cas9酶的质粒转染细胞后的第2天测得的试卤灵荧光的归一化倍数变化。左侧柱显示了用dmso处理的细胞的归一化倍数变化数,右侧柱显示了用pft
‑
α处理的细胞的归一化倍数变化数,pft
‑
α专门阻断肿瘤抑制因子p53的
转录活性。右侧柱显示的数字高于左侧柱,这意味着用pft
‑
α处理的细胞具有增加的试卤灵荧光测量值,因此pft
‑
α降低了细胞毒性。这表明在a549细胞中看到的与crispr
‑
cas9系统相关的细胞毒性至少部分依赖于p53,这是在其他人细胞类型中看到的驱动cas9介导的细胞毒性的主要因素。a549细胞对p53活性呈阳性。
[0141]
图6显示柱形图,其显示用具有不同grna序列的rnp质粒转染的细胞相比于对照细胞的试卤灵荧光的归一化倍数变化。图6的分图a显示了设计用于靶向hbb基因的外显子1的三个grna序列(g1、g2和g3)。在图6的分图b中,y轴显示归一化倍数变化数,x轴显示nt hbb
‑
g1、nt hbb
‑
g2、nt hbb
‑
g3和对照的列。对照的试卤灵荧光测量值归一化为1,因此,它们的归一化倍数变化数为1。nt hbb
‑
g3具有最小的归一化倍数变化数,这表明它具有最少的试卤灵荧光。图6的分图c显示了cas9 hbb
‑
g3反向序列迹线,表明indel的产生,将毒性与核酸酶切割活性联系起来。
[0142]
图7显示了实验设计的实施方案。在96孔板中培养细胞,每孔接种约2.5x104个细胞。96孔板的每一列分别接受不同质粒的处理。因为每列包含8个孔,每个处理有8个重复。第一列中的细胞用编码如图1所示的第一融合蛋白的质粒转染;第二列中的细胞用编码如图1所示的第二融合蛋白的质粒转染;第三列中的细胞用编码如图1所示的第三融合蛋白的质粒转染;第四列中的细胞用编码如图1所示的第四融合蛋白的质粒转染;第五列中的细胞用编码如图1所示的第五融合蛋白的质粒转染;第六列中的细胞用编码如图1所示的第六融合蛋白的质粒转染;第七列中的细胞用编码如图1所示的第七融合蛋白的质粒转染;第八列中的细胞用编码如图1所示的第八融合蛋白的质粒转染;以及第九列中的细胞用编码如图1所示的第九融合蛋白的质粒转染。此外,第十列中的细胞用编码未修饰的cas9酶的质粒转染,而第十一列中的细胞用编码未修饰的cas9酶的质粒以及编码hexo1(1
‑
352)的质粒转染。第十二列中的细胞用编码gfp的质粒转染。第十三列中的细胞是未经任何处理的对照。
[0143]
图8显示柱形图,其显示测得的试卤灵荧光的归一化倍数变化。y轴显示归一化倍数变化数,x轴显示使用分别编码9种融合蛋白和g3(seq id no:23)grna的rnp质粒的处理。x轴进一步显示了两个另外的处理:一个处理用编码未修饰的cas9和g3 grna(cas9 wt)的rnp质粒转染细胞;另一个处理用单独编码未修饰的cas9和hexo1以及g3 grna(cas9 wt+exo1)的rna质粒转染细胞。此外,x轴显示gfp处理组和未进行任何处理的对照组。对照组的试卤灵荧光测量值归一化为1,因为归一化倍数变化数为1。将每隔一个处理的试卤灵荧光测量值与对照组进行比较,以获得归一化倍数变化数。正如预期的那样,从所有其他处理中都观察到了不同程度的细胞毒性,其中cas9和cas9+hexo1组显示出最小的归一化倍数变化数,这表明来自两个阳性对照组的转染细胞具有最少量的试卤灵荧光,并另外证明了将hexo1与cas9直接融合以降低毒性的必要性。
[0144]
图9a
‑
b显示柱形图,其显示测得的试卤灵荧光的归一化倍数变化。图9a显示了靶向hbb基因的外显子1的g2和g3 grna。在图9b的分图中,y轴显示归一化倍数变化数,x轴显示使用编码第七融合蛋白与g3 grna的rnp质粒和编码未修饰的cas9和g2 grna的rnp质粒和对照的处理。对照组的试卤灵荧光测量值归一化为1,因为归一化倍数变化数为1。将每隔一个处理的试卤灵荧光测量值与对照组进行比较,以获得归一化倍数变化数。编码未修饰的cas9和g2 grna组的rnp质粒显示出最低的归一化倍数变化数,这表明来自该组的转染细胞具有最少量的试卤灵荧光。
[0145]
图10显示了在有或没有单链同源定向修复模板(hdrt)的情况下,用靶向hbb基因的外显子1的不同rnp质粒转染的细胞的试卤灵荧光的归一化倍数变化。在有和没有hdrt情况下,cas9
‑
hr7都显示出试卤灵荧光增加(因此细胞毒性降低)。此外,添加hdrt可降低cas9
‑
hr7和野生型cas9(nt)的毒性,但是我们不确定这种影响是否是特异性的(需要hbb外显子1的同源臂),或者hdrt是否只是与编码cas9
‑
hr7和cas9(nt)的质粒竞争转染。无论如何,cas9
‑
hr7显示出降低的毒性。
[0146]
图11a是质粒px330的示意图,其包含用于哺乳动物cas9表达的组成型启动子,以及驱动的grna表达的u6启动子。该质粒经过修饰以产生各种cas9
‑
hr版本1
‑
9。
[0147]
图11b是实验设置的示例,其中将细胞接种在96孔玻璃底孔板中。细胞毒性在转染后两天通过刃天青转化为试卤灵进行量化,然后将其针对未转染的对照进行归一化,以便跨实验进行准确比较。如上面96孔板所示,每列是不同的处理,8行为每个处理提供8个独立的重复。
[0148]
图11c显示了使用靶向12号染色体上的基因间区域的grna在a549细胞中降低了细胞毒性的图。cas9
‑
hr构建体1
‑
8在a549细胞中的毒性明显低于未修饰的cas9,如更高的归一化倍数变化值所示。将cas9
‑
hr 1
‑
8、cas9、cas9+hexo1、gfp和未转染对照(con)的平均值针对con平均荧光归一化。重要的是,cas9和hexo1的物理偶联对于降低毒性是必要的,因为与单独的cas9相比,cas9和hexo1的转染不降低毒性。所有实验均在重复的独立孔板中进行(总共16个重复),误差棒代表平均值的标准误差。
[0149]
图11d是显示用α
‑
pifithrin(10微摩尔)处理降低a549细胞中cas9诱导的细胞毒性的图。作为图5的扩展,可以看出添加dmso不会改变相对于单独转染cas9的毒性,表明pft
‑
α所见的效果是特异性的。
[0150]
图12a是嘌呤霉素抗性修复模板(rt)的图。它包含5’同源臂(5’)、强组成型病毒启动子(pcmv)、嘌呤霉素抗性基因(puro)、poly
‑
a序列(sc40pa)和3’同源臂(3’)。修复模板下方是指导物int
‑
g2和g3所靶向的基因组区域。修复模板旨在两个指导物序列的中间整合,从而防止进一步的cas9切割。整合位点位于6号染色体上h2b
‑
b和h2b
‑
a之间的基因间区域,这允许测试cas9
‑
hr在基因组的基因间和编码区域中发挥功能的能力。此外,两条链都是靶向性的,测试cas9
‑
hr与有义和反义方向的兼容性。
[0151]
图12b显示了用于量化a549细胞中hexo
‑
cas9融合物毒性的方法。a549细胞点种(plate)在96孔板中,通过如前所述的标准cal
‑
phos方案转染500ng的每种质粒和100ng的修复模板。
[0152]
图12c是描绘通过刃天青测定测试的各种构建体的毒性的图。所有cas9
‑
hr构建体都显示出比cas9
‑
nt显著更低的毒性(更高的归一化荧光),其中8个与仅有修复模板的对照相比在毒性上没有统计学显著差异。此外,靶向有义和反义的cas9
‑
hr显示出类似的毒性降低,表明cas9
‑
hr可以在任一取向发挥作用。
[0153]
图12d描绘了测量cas9
‑
hr8和cas9的hdr活性的测定方法。该测定通过测量用嘌呤霉素处理后的细胞存活率来确定hdr率。由于这是存活率测定,并且a549细胞对cas9的转染表现出显著的p53依赖性细胞毒性,因此改用k562细胞(p53
‑
/
‑
)以促进准确定量hdr率。在用500ng的cas9
‑
hr8或cas9和100ng的修复模板电穿孔后,将k562细胞等分到12孔板中。两天后提取dna并开始嘌呤霉素(0.5mg/ml)选择。选择三天后,通过刃天青在96孔板中对k562
细胞进行定量。
[0154]
图12e描绘了通过puro
‑
rt成功整合的细胞基因组区域。左引物对和右引物对被设计使得一个引物在修复模板外的基因组区域结合,而另一个结合特异于嘌呤霉素盒的序列。5’和3’引物组的成功扩增强烈表明转基因的正确整合。
[0155]
图12f显示了用具有g2或g3 grna的cas9
‑
hr8或cas9转染的k562细胞在嘌呤霉素处理三天后的存活数据。将数据针对用含有rt的质粒转染的细胞进行归一化。在嘌呤霉素选择3天后,靶向有义(int
‑
g2)和反义(int
‑
g3)的cas9
‑
hr8显示相对于野生型cas9(nt)归一化试卤灵荧光增加了超过两倍。增加两倍的试卤灵荧光转化为hdr率增加至少两倍,表明cas9
‑
hr不仅可以显著降低毒性,而且它们还可以增加多种细胞类型的hdr率和cas9
‑
hr功能。
[0156]
图12g显示了靶区域被5’引物对和3’引物对扩增后的琼脂糖凝胶,显示修复模板已经被的图1的第8融合蛋白和cas
‑
9对照(nt)成功整合。当仅使用gfp构建体(gfp)或不使用模板(con)转染时,靶区域没有扩增。
[0157]
图13a显示了基因组区域,包括hbb的前两个外显子,该hbb被靶向以编辑人血红蛋白β(hbb)基因。插图显示了外显子1的更大版本,其中图描述了测试的grna。该图显示了a549细胞中hbb grna指导物的毒性筛选数据。如图11d中那样进行毒性实验,其中hbb
‑
g3显示出比hbb
‑
g1或hbb
‑
g2更高的毒性。
[0158]
图13b显示hbb
‑
g3处理的a549细胞中hbb基因组区域的桑格测序。桑格测序显示cas9切割和通过nhej途径修复后的特征性噪音,其中条形指示grna序列。在这种情况下,由于具有反向引物的序列,噪音是5’而不是3’。在用hbb
‑
g1或hbb
‑
g2处理的细胞中无法检测到清晰切割和通过nhej的修复。
[0159]
图13c是野生型hbb序列和ssrt
‑
g3序列的示意图,ssrt
‑
g3序列向镰状细胞(e6v)引入了产生错义突变的ecori位点和四个沉默错配突变(粗体a、a、a和g核苷酸碱基),其中hbb
‑
g3 grna由上面的条突出显示。单链修复模板(ssrt)
‑
g3长120bp,其中在预测的切割位点两侧各有60bp臂。突变被设计为防止成功修复后grna结合。
[0160]
图13d描绘了hbb编辑实验,其中k562细胞或a549细胞用cas9+ssrt
‑
g3、cas9
‑
hr1
‑
9+ssrt
‑
g3或单独的ssrt
‑
g3进行电穿孔。两天后,细胞如图11d中被定量。然后提取dna,并用两个引物对扩增hbb基因座。外部对用ecori消化以量化hdr编辑率,内部对可用于深度测序,以提供除indel率之外的hdr率的独立量化,从而可以准确量化hdr/indel比率。
[0161]
图14图示了通过用hbb
‑
g3 grna和cas9
‑
hr融合蛋白4和5(如图1所示)转染a549细胞确定的两种转染方法lipofectamine和磷酸钙(calphos)的毒性评估。使用cal
‑
phos或lipofectamine看到的cas9
‑
hr4和5的类似结果强烈表明毒性效应不依赖于特定的转染试剂/方法。此外,cas9(nt)的lipofectamine转染显示出相对于cal
‑
phos转染的毒性增加,这表明与可能更有效的lipofectamine转染相比,cal
‑
phos转染实际上可能低估了cas9
‑
hr对毒性的降低。
[0162]
图15图示了通过用图13a的ssrt hbb修复模板转染a549细胞进行的毒性评估。在转染后第2天测量刃天青水平。cas9
‑
hr融合蛋白4和8在a549细胞中毒性较小。ssrt降低毒性细胞毒性,尤其是对于nt。
[0163]
图16a显示了描绘图1的cas9
‑
hr融合蛋白8的ecori消化测定的琼脂糖凝胶,该融
合蛋白将hbb修复模板整合到k562细胞的基因组中。箭头描绘ecori消化的产物。在仅cas9(nt)、ssrt和con(无cas9)的泳道中没有可检测到的ecori消化的产物。这表明cas9
‑
hr在修复模板选择方面具有灵活性,ssrt和双链(ds)rt均可用于基因组编辑。
[0164]
图16b显示了描绘图1的cas9
‑
hr融合蛋白4、5、6、7和8的ecori消化测定的额外琼脂糖凝胶,该融合蛋白将hbb修复模板整合到k562细胞的基因组中。箭头描绘了ecori消化的产品,表示成功的hdr。考虑到之前的毒性结果(图8),正如预期的那样,cas9
‑
hr4似乎具有最高的hdr率,与未转染对照(con)的消化相比时所有其他cas9
‑
hr和cas9(nt)均显示出一定程度的成功hdr。
[0165]
图16c显示了cas9
‑
hr融合蛋白4、5、6、7和8(如图1中所示),仅cas9(nt)和con(无cas9)的蛋白质印迹。箭头表示在cas9
‑
hr融合蛋白和nt泳道中检测到cas9。虽然对于融合物4
‑
7而言量似乎较低,但另外的印迹和ihc(图16e)显示所有cas9
‑
hr的正确表达和定位,表明毒性的降低不太可能是由于表达水平降低。例如,cas9
‑
hr4和8是通过蛋白质印迹法测定的cas9
‑
hr的一些最低和最高表达子。如果通过减少表达真正降低了细胞毒性,那么预期cas9
‑
hr4将在基因组中测试的每个靶处具有最低的毒性。然而,情况并非如此,如图11c和图12c表明cas9
‑
hr4实际上在所有测试的cas9
‑
hr中毒性最高,而cas9
‑
hr8毒性最低者之一。鉴于这些结果,表达水平更有可能在决定细胞毒性方面没有发挥重要作用,进一步证明了当靶向hbb exo1(图8)时,在所有测试的cas9
‑
hr中,cas9
‑
hr4的毒性降低幅度最大,因此显示表达水平和毒性之间没有明确的相关性。最后,有趣的是cas9
‑
hr4和8似乎在基因组的不同位点显示出互补的毒性降低:如果cas9
‑
hr4降低毒性,则cas9
‑
hr8不会(或效果较差),反之亦然。这可能与这些不同环境中的不同局部染色质环境有关,不同的接头身份可能允许hexo1结构域在不同染色质环境中的最佳定位。因此,使用不同版本的cas9
‑
hr可以在整个基因组几乎所有位置处降低毒性(并增加hdr)。
[0166]
图16d图示了cas9
‑
hr3从大肠杆菌的成功表达和纯化,通过sds
‑
page用考马斯染色监测。泳道l是梯度。泳道1和8是细胞裂解物的可溶性级分。泳道2和9是不溶性裂解细胞沉淀。泳道3和10是通过镍(ni
‑
nta)柱的可溶性级分的流穿液(flow
‑
through)。泳道4和11是洗脱级分,其中与镍结合的蛋白质被洗脱。泳道5和12是磺酰基(sp)阳离子交换色谱树脂的流穿液。泳道6和13是用500mm nacl洗脱的洗脱级分。泳道7和14是用1m nacl洗脱的洗脱级分。泳道1
‑
7来自用cas9
‑
hr3转染的细胞。泳道8
‑
14作为纯化方案的对照,来自仅表达未修饰的cas9的大肠杆菌。为cas9
‑
hr开发成功的基于大肠杆菌的蛋白质纯化方案允许cas9
‑
hr活性的体外测试,以及各种真核生物的直接rnp转染和编辑。
[0167]
图16e显示了来自图16c的相同转染细胞的免疫组织化学(ihc)。箭头表示cas9
‑
hr融合物和cas9定位于细胞核。细胞核中所有cas9
‑
hr4
‑
8的检测和正确定位(5
‑
7测定和正确定位可见,数据未显示)进一步证明cas9
‑
hr对毒性的降低不是由于定位不当,也不是由于通过ihc测定的表达水平的显著降低。
[0168]
图17a图示了用于h2bmneon敲入实验的修复模板的设计。该实验允许在基于非生存的测定中通过正确定位的gfp荧光准确量化hdr率。
[0169]
图17b图示了由图1的cas
‑
hr融合蛋白4、5、6和8,仅cas9(nt)和con(无cas9)在上皮肺癌细胞系中诱导的细胞毒性的p53依赖性降低。a549细胞对p53活性呈阳性,而h1299细胞对p53活性呈阴性。由归一化刃天青水平(y轴)确定的毒性表明,h1299细胞中p53的缺失
末端之外结合,与仅存在于基因组中而不存在于rt中的序列退火,而其他则与修复模板的特异性序列退火,并且不存在于未修饰的细胞中。5’和3’引物组的成功扩增强烈表明用mneon成功且正确地标记了h2b。
[0176]
图19b图示了琼脂糖凝胶,其显示了从用cas9
‑
hr4、8和cas9nt加h2bmneon rt以及未转染的对照(泳道4、8、nt和con)转染的k562细胞提取的gdna扩增的pcr产物。使用来自cas9
‑
hr4、8和cas9(nt)的5’引物和gdna的扩增均显示5’产物的成功扩增,而con则没有,表明rt的5’末端的正确整合。另外,使用来自cas9
‑
hr8的gdna的更高量的扩增产物对应于图17c中看到的更高的hdr率。
[0177]
图19c,从用cas9
‑
hr4、8和cas9nt加h2b
‑
mneon rt以及未转染的对照(泳道4、8、nt和con)转染的k562细胞提取的gdna扩增的pcr产物。虽然cas9
‑
hr8和cas9的水平看起来相似,但鉴于这两者的扩增明显更高,反应很可能已经超过了指数期,使得量化不太可靠。不管怎样,使用来自cas9
‑
hr4、8和cas9(nt)的3’引物和gdna的扩增均显示3’产物的成功扩增,而con泳道未显示特定条带,表明rt的3’末端的正确整合。
[0178]
图19d图示了由5’引物从cas9
‑
hr8扩增的pcr产物的桑格测序序列迹线的吸光度。顶部迹线显示产物的5’序列,其中白色条显示仅存在于基因组中的序列,而阴影条显示同时存在于rt和基因组中的序列。间插序列被裁剪掉,底部的迹线显示了产物的3’末端。阴影条再次代表h2b orf,而白色条代表mneon。此外,阴影条显示了为防止转基因整合后的另外切割而引入的两个沉默突变。cas9
‑
hr4和cas9(nt)迹线是相同的。
[0179]
图19e图示了由3’引物从cas9
‑
hr8扩增的pcr产物的桑格测序序列迹线的吸光度。顶部迹线显示产物的5’序列,其中白色条显示mneon,而阴影条显示同时存在于rt和基因组中的序列。间插序列被裁剪掉,底部的迹线显示了产物的3’末端。阴影条再次代表h2b 3’区域,虚线显示从基因组和rt到仅基因组序列的转变。此外,三个箭头显示了相对于参考序列的snp。cas9
‑
hr4包含类似的突变,而cas9迹线在rt的末端后立即降解。这些更有可能代表键化(bonified)的snp,但不能排除cas9
‑
hr可能会在连接位点周围引起一些错误。对照细胞的直接测序将有助于解决这个问题。
[0180]
图19f图示了由5’引物扩增的pcr产物的测序比对。未发现相对于预期参考序列的错误。
[0181]
图19g图示了由3’引物扩增的pcr产物的测序比对。相对于预期序列的唯一变化出现在rt序列之外,并且很可能显示了相对于参考序列的细胞系特异性snp。
[0182]
图20图示了具有通过至少两个接头连接到cas9的结构域组合的另外的cas9
‑
hr融合蛋白。这些不同的融合物可能会增加hdr率和/或进一步降低细胞毒性。实施例
[0183]
给出以下实施例的目的是为了说明本公开中描述的各种实施方案,并不意味着以任何方式进行限制。本实施例连同在此描述的方法目前代表优选实施方案,是示例性的,并不意在限制本公开的范围。本领域技术人员将想到包含在由权利要求的范围限定的本公开的精神内的变化和其他用途。实施例1
–
a549细胞中降低的细胞毒性
[0184]
参考图1,产生了几种具有不同多核苷酸的质粒构建体。每个多核苷酸编码不同的融合蛋白,所述融合蛋白由通过特定的接头肽连接到cas9(seq id no:2
‑
18的任何一个)的
hexo1片段(seq id no:1的氨基酸1
‑
352)构成。一些质粒构建体编码具有一个n末端核定位序列(nls)或c末端nls的cas9酶,而一些质粒构建体编码具有c末端nls和n末端nls的cas9酶。对所有质粒构建体进行测序以确保多核苷酸序列中不发生突变。每个质粒构建体还包含编码指向预期染色体位点的grna的核苷酸序列。预期的染色体位点位于12号染色体的5’上的vsp33a和3’上的clip1之间的基因间区域中,该区域没有预测的基因或长的非编码rna。一旦用质粒转染细胞,细胞内就会形成cas9
‑
grna核糖核蛋白(rnp)。制备对照质粒以编码未修饰的cas9(seq id no:2
‑
18中的任一个)酶。
[0185]
培养人肺癌a549细胞并将约2.5x104个细胞点种在96孔板中,其中8
‑
16个转染重复/单个处理。然后使用标准磷酸钙转染技术用62.5ng质粒dna转染每个孔,并孵育过夜16
‑
20小时。然后让细胞恢复一天。刃天青还原测定(图3)用于估计96孔板中活细胞的数量。刃天青是一种可渗透细胞的氧化还原指示剂,可用于监测活细胞数量。刃天青可以溶解在生理缓冲液中(产生深蓝色溶液),并以均质形式直接添加到96孔板中的培养物中的细胞中。代谢活跃的活细胞可将刃天青还原成呈粉红色荧光的试卤灵产物。此外,产生的试卤灵的数量与活细胞的数量成正比,可以使用配备535nm激发/590nm发射滤光片组的微孔板荧光计进行量化。
[0186]
参考图4,在质粒dna转染后两天,与用编码未修饰cas9酶的对照质粒转染的细胞相比,大多数用编码融合hexo1
‑
cas9蛋白的质粒转染的细胞在统计学上增加了细胞活力(约3
‑
4倍)。此外,用hdr模板、对照抗体或gfp质粒处理的细胞和未接受处理的细胞具有相似的细胞活力。实施例2
–
使用靶向hbb基因的grna降低a549细胞中的细胞毒性
[0187]
与实施例1中进行的实验类似,产生了几个含有编码融合蛋白hexo1
‑
cas9酶的多核苷酸的质粒(图1)。每个质粒构建体还包含编码用于识别人hbb基因的外显子1的grna的核苷酸序列。与实施例1中进行的实验相比,引入了用具有分别编码野生型cas9酶的核苷酸序列和编码hexo1的核苷酸序列的质粒转染细胞的另外对照。使用了表2中列出的用于识别hbb基因的外显子之一的三个grna序列。制备对照质粒以编码未修饰的cas9(seq id no:2
‑
18中的任一个)酶。
[0188]
使用与实施例1中进行的实验类似的细胞培养和转染方案。参考图6,与grna g1(seq id no:21)和grna g2(seq id no:22)相比,grna g3(seq id no:23)具有最高的细胞毒性。参考图8,与使用两种对照处理的细胞相比,用rnp质粒转染的细胞通常具有更高的活细胞百分比。此外,图9b显示了与具有未修饰的cas9和g3 grna的rnp质粒相比,具有第七融合蛋白(图1)和g2 grna的rnp质粒具有更小的细胞毒性。实施例3
‑
治疗患者的镰状细胞性贫血
[0189]
从患有镰状细胞性贫血的受试者获得生物样品。从生物样品中提取基因组dna并测序以验证β
‑
珠蛋白基因的氨基酸6密码子中的单核苷酸取代(a到t)。该突变将谷氨酸密码子(gag)转化为缬氨酸密码子(gtg)。从患者的骨髓腔中分离出造血干细胞并进行离体培养。编码hexo1
‑
cas9的蛋白质融合复合物和grna部分的核酸载体被递送到培养的造血干细胞中。此外,将带有编码β
‑
珠蛋白基因外显子1野生型序列的整合盒的dna模板序列递送到培养的造血干细胞中。grna部分包含与β
‑
珠蛋白基因外显子1的gtg基因座互补的至少10个核苷酸。dna模板序列包含侧接有5’同源区和3’同源区的整合盒,其中5’同源区和3’同源区
与外显子1的gtg基因座侧翼的区段表现出至少80%的同一性。多核苷酸的整合盒包含野生型gag序列,该序列对应于在原代细胞中检测到的染色体异常基因座。在将编码rnp和dna模板序列的核酸递送到培养的造血干细胞中后,grna将工程改造的hexo1
‑
cas9蛋白引导至gtg基因座,在那里工程改造的hexo1
‑
cas9蛋白的cas9部分产生dsb。工程改造的hexo1
‑
cas9蛋白的hexo1部分部分地消化了切割的gtg基因座,留下了3’突出端。dna模板序列的存在通过hdr促进内源性修复,其中在β
‑
珠蛋白基因外显子1的氨基酸6处具有正确野生型序列gag的整合盒被插入造血干细胞的染色体中。筛选出具有校正的gag序列的造血干细胞并选择将其移植回患者体内。实施例4
–
使用靶向12号染色体基因间区域的grna降低a549细胞中的细胞毒性
[0190]
与实施例2中进行的实验类似,产生了几个含有编码融合蛋白hexo1
‑
cas9酶(图11a和图1)的多核苷酸的质粒。每个质粒构建体还包含编码grna的核苷酸序列,该序列用于识别12号染色体上的基因间区域,其中该染色体的a549细胞具有两个拷贝。与实施例3中进行的实验相比,还引入了用具有分别编码野生型cas9酶的核苷酸序列和编码hexo1的核苷酸序列的质粒转染细胞的对照。制备对照质粒以编码未修饰的cas9(seq id no:2
‑
18中的任一个)酶。
[0191]
使用与实施例2中进行的实验类似的细胞培养和转染方案。将大约2.5*10^4个细胞点种在96孔板中,其中8
‑
16个重复/单独实验,如图11b所示。参考图11c,与使用两种对照处理的细胞相比,用px330质粒转染的细胞通常具有高得多的活细胞百分比,细胞活力增加了3
‑
4倍。图11c还表明hexo1的融合导致细胞毒性降低,因为cas9和hexo1的共表达不影响细胞毒性。图11d表明,用αpfithrin对转染了野生型cas9的细胞进行处理降低了由cas9的活性引起的毒性。图11d中所示的cas9失活表明在a549细胞中cas9处理引起毒性的原因至少部分是由于基于细胞凋亡的p53激活,与ihrey等人和haapaniemi等人的研究相同。实施例5
–
a549细胞中hexo
‑
cas9融合物的hdr和indel率的量化
[0192]
cas
‑
9hexo1融合物用于将抗生素抗性盒整合到a549细胞6号染色体上的基因座中。嘌呤霉素抗性修复模板如图12a所示。它包含5’同源臂(5’)、强组成型病毒启动子(pcmv)、嘌呤霉素抗性基因(puro)、poly
‑
a序列(sv40pa)和3’同源臂(3’)。修复模板下方显示了指导物int
‑
g2和g
‑
3所靶向的基因组区域。修复模板旨在破坏两个指导物序列的中间的整合,从而防止进一步的cas9切割。整合的成功通过抗生素选择来量化。a549细胞只有6号染色体的一个拷贝。靶整合位点距离6号染色体上人h2b基因的3’末端约1kb。该区域没有预测的基因。
[0193]
使用与实施例4中进行的实验类似的细胞培养和转染方案。将大约2.5*10^4个细胞点种在96孔板中,其中8
‑
16个重复/单独实验,如图12b所示。
[0194]
图12c显示,与其他融合蛋白和cas9相比,分别具有g2 grna和g3 rna的cas9
‑
hr8在第2天显示出最高的a549细胞存活率。由于这个结果,cas9
‑
hr8被用在以下实施例中。实施例6
–
k562细胞中hexo
‑
cas9融合物的hdr和indel率的量化
[0195]
与实施例5中的实验相比,使用k562细胞并使用neon(thermofisher)电穿孔。k562细胞缺乏p53功能。根据实施例5的结果,重要的是通过cas9的活性去除p53激活的变量,因为这在融合物cas9和野生型cas9之间会有所不同,从而引入了影响抗生素筛选结果的可能性。
[0196]
k562细胞用500ng的每个质粒和100ng的修复模板进行电穿孔,如图12d所示。两天后,从大约1/10的存活细胞中提取dna,用于分析puro rt基因组整合。第二天,加入0.5mg/ml嘌呤霉素,三天后用标准刃天青测定法对细胞存活进行量化,如图12d所示。
[0197]
如实施例5中那样进行毒性定量,其中加入融合构建体9。图12f显示与cas9(具有grna g2和g3)相比,cas9
‑
hr8(具有grna g2和g3)之间的细胞毒性显著降低,存活细胞的数量增加了一倍。
[0198]
使用基因组特异性引物和修复模板特异性引物的成功扩增表明修复模板的成功整合,并且cas9
‑
hr系列构建体的毒性降低不是由于缺乏核酸酶活性。可能有迹象表明cas9
‑
hr系列的编辑效率高于cas9。
[0199]
图12e显示了成功整合修复模板的细胞的基因组区域的描述。在第2天对转染的细胞进行定量。从每个处理的一个孔中提取dna。用1微克/毫升嘌呤霉素处理7天后,用刃天青对细胞进行定量。从另一排细胞中提取dna。插入连接点用左右引物对扩增。可以执行深度测序来鉴定具有成功hdr的细胞中的indel率。来自第2天的dna用于通过用左右引物扩增来量化hdr细胞中的indel率,如图12e所示。
[0200]
图12g描绘了扩增产物上的凝胶电泳结果,其显示用cas9
‑
hr8(8)或具有grna g2或g3(nt)的cas9转染的k562细胞成功产生具有图12e中描绘的两个引物对的扩增子,而gfp或转染的细胞没有。这表明修复模板已成功整合。实施例7
–
确定毒性和cas9活性之间的关系
[0201]
如实施例2中所见,不同的指导rna可具有完全不同的切割率和毒性。使用与实施例4相同的方法将具有未修饰的cas9和靶向图13a中所示区域的指导物的构建体转染到a549细胞中。如实施例1中那样使用刃天青量化毒性。
[0202]
从用hbb
‑
g1、hbb
‑
g2和hbb
‑
g3转染的细胞中提取dna,用图13d中的外引物对进行扩增并发送用于桑格测序。只有hbb
‑
g3显示出显著的切割,这由图13b中所示切割位点后噪声的特征性增加所证明。这表明毒性是a549细胞中cas9核酸酶活性的良好代表。因此在实施例8中使用了指导rna hbb
‑
g3。实施例8
‑
使用cas9
‑
hr编辑已知疾病基因座
[0203]
与实施例7类似,使用k562细胞是因为它们缺乏p53活性以及因为它们与血细胞的相似性比a549细胞更高。
[0204]
实施例7的grna——hbb
‑
g3分别与cas9和cas9
‑
hr 1
‑
9转染以将多个突变引入k562细胞的hbb基因座中。第一突变选择是镰状细胞e6v突变。镰状细胞e6v突变与另外的突变一起产生,创建ecori限制性位点和两个沉默突变,旨在防止修复模板一旦整合到基因组中被重新切割,此外在预测的切割位点的每一侧都有60bp同源臂。
[0205]
通过电穿孔实现转染。电穿孔后两天,如实施例6用刃天青进行毒性测定。还收获了dna并扩增了hbb基因座,以准备进行深度测序以测量indel和hdr率。可替代地,可以用ecori消化dna以测量靶效率。图16a和图16b图示了在整合修复模板后,基因组基因座现在可以用ecori消化。ecori消化的扩增子可以在cas9
‑
hr4、cas9
‑
hr5、cas9
‑
hr6、cas9
‑
hr7和cas9
‑
hr8泳道中观察到。图16c、图16d和16e证实cas9
‑
hr表达并定位于转染的细胞的细胞核。实施例9
‑
编辑cd34+造血干细胞
[0206]
对cd34+细胞重复实施例8的实验。实施例8的grna——hbb
‑
g3分别与cas9和cas9
‑
hr1
‑
9转染以将多个突变引入k562细胞的hbb基因座中。第一突变选择是镰状细胞e6v突变。镰状细胞e6v突变与另外的突变一起产生,创建ecori限制性位点和两个沉默突变,旨在防止修复模板一旦整合到基因组中被重新切割,此外在预测的切割位点的每一侧都有60bp同源臂。
[0207]
通过电穿孔实现转染。电穿孔后两天,如实施例6用刃天青进行毒性测定。收获了dna并扩增了hbb基因座,以准备进行深度测序以测量indel和hdr率。可替代地,用ecori消化dna以测量靶效率。实施例10
‑
cas9
‑
hr3的体外核酸酶活性
[0208]
使用标准taq dna聚合酶和hbb
‑
out
‑4‑
f(5
’‑
aacgatcctgagacttccaca
‑3’
(seq id no:127))和hbb
‑
out
‑5‑
r(5
’‑
tgcttaccaagctgtgattcc
‑
3'(seq id no:128)),tm=56,35个周期,从野生型k562细胞中扩增出一段954bp的dna,并使用qiagen pcr纯化试剂盒进行纯化。接下来,将hbb
‑
g1(5
’‑
guaacggcagacuucuccuc
‑3’
(seq id no:129),idt)或hbb
‑
g3(5
’‑
gaggugaacguggaugaagu
‑3’
(seq id no:130),idt)与tracrrna(idt)在双链体缓冲液(idt)中以各自终浓度为1μm进行组合。将rna在95℃下加热5分钟,然后冷却至室温。然后将cas9或cas9
‑
hr3与hbb
‑
g1或hbb
‑
g3指导rna复合物组合,并在1x cas9反应缓冲液(50mm tris,100mm nacl,10mm mgcl2,1mm dtt,ph 7.9)中以10:10:1摩尔比(30nm:30nm:3nm)扩增dna并在37℃下孵育1小时,然后加入1μl蛋白酶k,并将反应在50℃下再孵育20分钟。然后将样品在标准的1%tae琼脂糖凝胶上进行电泳并成像。
[0209]
图18a图示了cas9
‑
hr的机制建模。cas9与预定位点结合,切割,然后保持结合状态,直到被蛋白酶k消化掉。由于cas9
‑
hr具有另外的5
’‑
>3’外切核酸酶活性,预计会出现更复杂的模式。重要的是,已表明hexo1对磷酸化的5
’‑
双链dna末端的亲和力大约是未磷酸化的10倍。这会导致两个重要的后果。首先,预计会在不添加任何grna的情况下对pcr进行一些小的消化,这通常预计不会发生在cas9的情况下。改变用于扩增dna片段(带有5
’‑
磷酸酯键或硫酯键)的引物的性质可以分别增加或减少这种降解。其次,由于双链dna的切割(dsdna)产生具有5
’‑
磷酸酯的末端,预计原始cas9
‑
hr或其他未结合的cas9
‑
hr分子会以5
’‑
>3’切除dsdna,生成各种dsdna、双链和单链(ds::ss)dna和ssdna产物的混合。图18b图示了基于图18a的机制的预期cas9和cas9
‑
hr消化模式。图18c图示了图18a和图18b的实际琼脂糖示例。泳道1和2显示cas9
‑
hr3靶向hbb
‑
g1或hbb
‑
g3,泳道3和4显示cas9(nt)靶向hbb
‑
g1或hbb
‑
g3,泳道5是未处理的dna。图18d图示了与图18c类似的实验,与图18b的差异在于将酶在4℃下放置2周后进行实验以比较蛋白质稳定性。泳道1是来自cas9
‑
hr3和grna hbb
‑
g1组合的消化模式。泳道2是来自cas9和grna hbb
‑
g1组合的消化模式。泳道3是来自cas9
‑
hr3和hbb
‑
g3组合的消化模式。泳道4是来自cas9和hbb
‑
g3组合的消化模式。泳道5是来自仅cas9
‑
hr的消化模式。泳道6是来自仅cas9的消化模式。泳道7是既没有cas9也没有grna的对照。图18c和图18d证明消化模式对应于如图18a和图18b所示的机制。实施例11
‑
hh2b基因组整合和基因组验证
[0210]
cas9
‑
hr和cas用于将hh2b片段引入h2b基因组基因座。引物被设计成使得基因组引物在h2b
‑
mneon修复模板(rt)之外,而另一个是rt特异性的(在mneon内),如图19a所示。3’引物的序列是h2b
‑
rt
‑3’‑
f:5
’‑
aggcctttaccgatgtgatg
‑3’
(seq id no:131),h2b
‑
rt
‑
3
’‑
r:5
’‑
acggagtctcgctctgtcac
‑3’
(seq id no:132)。5’引物的序列是h2b
‑
rt
‑5’‑
f:5
’‑
caaactgcaaggctgcaata
‑3’
(seq id no:133),h2b
‑
rt
‑3’‑
r:5
’‑
gacccaccatgtcaaagtcc
‑3’
(seq id no:134)
[0211]
转染k562细胞后,从用修复模板(rt)和cas9
‑
hr4、cas9
‑
hr8、cas9(nt)转染的细胞中或未转染(con)的细胞中提取基因组dna。使用标准taq聚合酶(bioneer,tm=56,35个周期)扩增5’引物或3’引物侧翼的片段。
[0212]
图19b图示了琼脂糖凝胶,其显示了由5’引物扩增的pcr产物。检测到cas9
‑
hr4、8和cas9
‑
nt的扩增产物,但未在未转染的对照中检测到。
[0213]
图19c图示了琼脂糖凝胶显示在cas9
‑
hr4、cas9
‑
hr8和仅cas9(nt)中3’引物的成功特异性扩增。
[0214]
图19d图示了由5’引物扩增的cas9
‑
hr8的pcr产物的桑格测序的序列迹线的吸光度。调用的碱基下方的实心或未填充的条表示dna的身份(左上方没有填充的条是基因组,中间两条带条纹的条是h2b orf,右下方没有填充的条是mneon),图19a的垂直灰色虚线显示rt中包含的基因组序列与单独的内源基因组序列之间的连接。从h2b到rt中包含的mneon序列到单独的内源基因组序列的清晰转变表明转基因在5’末端的成功整合。
[0215]
图19e图示了由3’引物扩增的cas9
‑
hr8的pcr产物的桑格测序的序列迹线的吸光度。调用的碱基的上方的条表示dna的身份(未填充的条是mneon,阴影的条是基因组)。从mneon到rt中包含的基因组序列到单独的内源基因组序列的清晰转变表明转基因在3’末端的成功整合。
[0216]
图19f和图19g图示了来自cas9
‑
hr4、cas9
‑
hr8和nt的5’(图19f)和3’(图19g)pcr产物的测序结果与参考序列的比对。使用clustalomega比对序列。实施例12
‑
编辑脂肪或脂肪前组织以增加代谢通量
[0217]
从患者分离未分化或成熟脂肪组织的细胞,并用编码任一版本的cas9
‑
hr或纯化的rnp的质粒转染。所选择的一个或多个cas9
‑
hr可以靶向人基因组的位点,该位点已被示出适合dna插入(“安全港位点”)或任何此类已识别的新位点。此外,同时转染包含解偶联蛋白(ucp)1、2、3的cdna的修复模板。该转基因包含所选整合位点的5’同源臂(ha),与基础启动子复合的泛增强子或组织特异性增强子,带有或不带有5’utr序列,由上述来自ucp 1、2或3的cdna组成的orf,带有或不带有3’utr序列,聚腺苷酸化序列,和所选的整合位点的3’ha。整合和随后重新引入表达这种转基因的脂肪组织可以增加基础代谢,导致整体体重减轻和脂肪脂质沉积物大小的减少。使用cas9
‑
hr可以降低毒性并增加成功整合的细胞的数量。实施例13
‑
编辑人真皮细胞以减少雄激素性脱发
[0218]
编码一个或多个cas9
‑
hr或纯化rnp的质粒可用于转染分离的细胞或在头皮上原位转染表达全长或经修饰的性结合激素球蛋白(sbhg)、nrf2或srd5a1、2或3的转基因。所选择的一个或多个cas9
‑
hr可以靶向人基因组的位点,该位点已被示出适合dna插入(“安全港位点”)或任何此类已识别的新位点。这些转基因包含所选整合位点的5’同源臂(ha),与基础启动子复合的泛增强子或组织特异性增强子,带有或不带有5’utr序列,由上述来自sbhg、nfr2或srd5a1、2或3的cdna组成的orf,带有或不带有3’utr序列,聚腺苷酸化序列,和所选的整合位点的3’ha。成功转染原位细胞或重新引入分离的真皮细胞可以延迟或永久停
止脱发并导致头发再生。
[0219]
虽然已经示出和在本文中描述了本公开的优选实施方案,对本领域技术人员将是显而易见的是,仅通过举例的方式来提供这样的实施方案。在不脱离本公开的情况下,本领域技术人员现在将想到许多变型、改变和替代。应当理解,可以采用本文所述的实施方案的各种替代方案。所附权利要求旨在限定本公开的范围,并且由此覆盖这些权利要求的范围内的方法和结构及其等同物。
[0220]
参考文献1.oakes,b.l.,nadler,d.c.&savage,d.f.protein engineering of cas9 for enhanced function.methods enzymol.546,491
–
511(2014).2.cong,l.et al.multiplex genome engineering using crispr/cas systems.science 339,819
–
823(2013).3.ran,f.a.et al.genome engineering using the crispr
‑
cas9 system.nat.protoc.8,2281
–
2308(2013).4.jinek,m.et al.a programmable dual
‑
rna
‑
guided dna endonuclease in adaptive bacterial immunity.science 337,816
–
821(2012).5.eid,a.,alshareef,s.&mahfouz,m.m.crispr base editors:genome editing without double
‑
stranded breaks.biochem.j.475,1955
–
1964(2018).6.gehrke,j.m.et al.an apobec3a
‑
cas9 base editor withminimized bystander and off
‑
target activities.nat.biotechnol.(2018).doi:10.1038/nbt.41997.wang,l.et al.in vivo delivery systems for therapeutic genome editing.int.j.mol.sci.17,(2016).8.zhang,j.
‑
p.et al.efficient precise knockin with a double cut hdr donor after crispr/cas9
‑
mediated double
‑
stranded dna cleavage.genome biol.18,35(2017).9.li,h.et al.design and specificity of long ssdna donors for crispr
‑
based knock
‑
in.biorxiv 178905(2017).doi:10.1101/17890510.canny,m.d.et al.inhibition of 53bp1 favors homology
‑
dependent dna repair and increases crispr
‑
cas9 genome
‑
editing efficiency.nat.biotechnol.36,95
–
102(2018).11.liang,x.,potter,j.,kumar,s.,ravinder,n.&chesnut,j.d.enhanced crispr/cas9
‑
mediated precise genome editing by improved design and delivery of grna,cas9 nuclease,and donor dna.j.biotechnol.241,136
–
146(2017).12.ihry,r.j.et al.p53 inhibits crispr
‑
cas9 engineering in human pluripotent stem cells.nat.med.24,939
–
946(2018).13.haapaniemi,e.,botla,s.,persson,j.,schmierer,b.&taipale,j.crispr
–
cas9 genome editing induces a p53
‑
mediated dna damage response.nat.med.24,927
–
930(2018).14.bieging,k.t.,mello,s.s.&attardi,l.d.unravelling mechanisms of p53
‑
activation and repression.mol.cell.biol.35,3800
–
9(2015).32.chang,h.h.y.,pannunzio,n.r.,adachi,n.&lieber,m.r.non
‑
homologous dna end joining and alternative pathways to double
‑
strand break repair.nat.rev.mol.cell biol.18,495
–
506(2017).33.jia,p.
‑
p.et al.role of human dna2(hdna2)as a potential target for cancer and other diseases:a systematic review.dna repair 59,9
–
19(2017).34.orans,j.et al.structures of human exonuclease 1 dna complexes suggest a unified mechanism for nuclease family.cell 145,212
–
23(2011).35.xiaoying,c.,jennica,z.&wei
‑
chiang,s.fusion protein linkers:property,design and functionality.adv drug deliv rev 65,1357
–
1369(2014).36.k.,engelhardt,h.,ringgeler,s.&h
ü
bner,h.basic colorimetric proliferation assays:mtt,wst,and resazurin.in cell viability assays:methods and protocols(eds.gilbert,d.f.&friedrich,o.)1
–
17(springer new york,2017).doi:10.1007/978
‑1‑
4939
‑
6960
‑
9_137.lieber,m.,todaro,g.,smith,b.,szakal,a.&nelson
‑
rees,w.a continuous tumor
‑
cell line from a human lung carcinoma withproperties of type ii alveolar epithelial cells.int.j.cancer 17,62
–
70(1976).38.klein,e.et al.properties of the k562 cell line,derived from a patient with chronic myeloid leukemia.int.j.cancer 18,421
–
431(1976).
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1