选择性雌激素受体降解剂的制作方法
选择性雌激素受体降解剂
1.相关申请
2.本技术要求2019年7月22日提交的印度临时专利申请号201921029554的优先权,其内容特此通过引用整体并入。
技术领域
3.本发明涉及一种选择性雌激素受体降解剂(serd),即化合物3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇和它的s对映异构体(2s)-3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇或其药学上可接受的盐。本发明进一步提供了它们的制备方法。本公开内容还涉及这些化合物用于治疗与雌激素受体(er)的调节相关的疾病(诸如er-阳性的乳腺癌)的用途和相关方法。
背景技术:
[0004]
通过与雌激素受体(er)的相互作用,内源性雌激素17β-雌二醇(e2)在生殖系统、骨骼发育和周转、心血管系统以及中枢神经系统中显示出多种生物活性。已发现er具有两种异形体er-α和er-β。已经充分确立雌激素和乳腺癌的生长和发展之间的联系。
[0005]
许多抑制内源性雌激素在雌激素受体阳性的乳腺癌中作用的策略正在实践中。这些包括:选择性的er调节剂(serm)诸如他莫昔芬,其作为乳房中er的选择性组织特异性拮抗剂;选择性的er降解剂(serd)诸如氟维司群,其促进er周转;和芳香酶抑制剂(ai)诸如依西美坦(甾体类)、阿那曲唑和来曲唑(非甾体类),它们抑制雌激素生物合成,并主要用于具有er-阳性的乳腺癌的绝经后妇女。不幸的是,许多患有乳腺癌的女性最初对他莫昔芬或ai疗法较好应答,但在治疗过程中在一段时间后产生抗性。在乳腺癌的抗性形式中,有证据表明,在雌激素受体下游的促生长信号传递途径仍然发挥着重要作用。最近,越来越多的临床证据表明,在用ai治疗后,由于er-α的配体结合结构域中的突变而产生抗性,使其即使在没有配体存在下也具有组成性活性,从而导致抗性。
[0006]
fanning报告说,最普遍的er-α点突变是y537s和d538g,而几个其它突变以显著降低的频率被鉴定。重要的是,基于乳腺癌细胞的研究表明,y537s和d538g突变赋予了er-α的激素非依赖性激活,并降低了临床上开处方的serm和serd的抑制效能和效力(fanning等人.elife 2016;5:e12792)。
[0007]
目前,氟维司群被认为是一流的serd。不幸的是,氟维司群的重大药学负担,即它对大体积肌肉内注射的需要、它的差溶解性以及它的口服生物利用度的缺乏,限制了它的广泛应用。因此,口服生物可利用的er拮抗剂、尤其是具有er降解特性的拮抗剂的开发将有益于对目前可得到的靶向er活性的疗法产生抗性的患者。最近已经开发了几种新型serd,它们目前处于不同的临床试验阶段,例如,sar-439859(ii期)、lsz-102(i期)、azd-9496(ii期)、gdc-810(目前停止)、gdc-927(目前停止)等。在现有技术中报道了许多非甾体类er拮抗剂。例如,美国专利号us5395842和us6060503公开了抗雌激素的化合物和组合物。
[0008]
美国专利号5,389,646、5,407,947、欧洲专利号0 470 310b1和wipo公开号wo 99/02512a1公开了苯并吡喃化合物,其可用于治疗或预防通过雌激素受体调节的病症。
[0009]
美国专利申请公开号2004/0034017(指定给schering aktiengesellschaft)公开了4-氟烷基-2h-苯并吡喃衍生物作为抗雌激素剂。
[0010]
wipo公开号wo 2011/156518a2和wo 2013/090829a1(二者指定给aragon pharmaceuticals inc.)公开了一大类2h-色烯衍生物作为雌激素受体调节剂。
[0011]
wo 2013/090836a1(指定给aragon pharmaceuticals,inc.)公开了在侧链中具有氟代氮杂环丁烷或吡咯烷环的2h-色烯衍生物作为雌激素受体调节剂/er降解剂。
[0012]
wo 2014/205136a1和wo 2014/205138a1(二者指定给seragon pharmaceuticals,inc.)公开了在侧链中具有氟甲基氮杂环丁烷基团的4-甲基-2h-色烯衍生物及其立体异构体作为雌激素受体调节剂/er降解剂。
[0013]
wo 2016/097073a1(指定给f.hoffmann-la roche ag/genentech,inc.)公开了在侧链中具有氟甲基氮杂环丁烷基团或氟甲基吡咯烷基团的2h-色烯衍生物作为雌激素受体调节剂/er降解剂。
[0014]
wo 2016/189011a1(指定给f.hoffmann-la roche ag/genentech,inc.)公开了在侧链中具有氟甲基氮杂环丁烷或吡咯烷基团的2h-色烯衍生物作为雌激素受体调节剂/er降解剂。
[0015]
wo 2013/090921a1和wo 20142/03132a1(二者指定给olema pharmaceuticals,inc.)公开了在侧链中具有甲基吡咯烷的苯并吡喃衍生物作为抗雌激素剂。
[0016]
wo 2016/174551a1(指定给pfizer inc.)公开了在侧链中具有n-烷基化的氮杂环丁烷的2h-色烯衍生物作为抗雌激素剂。
技术实现要素:
[0017]
本发明提供了由式i表示的化合物3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇:
[0018][0019]
或其药学上可接受的盐。
[0020]
本发明也提供了由式ia表示的化合物(2s)-3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇:
[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇:
[0031][0032]
或其药学上可接受的盐。
[0033]
当本发明的发明人将式i的化合物手性拆分为其对映异构体时,令人惊讶地发现,就其在mcf-7生长抑制测定/er-α降解测定中的体外效能以及其药代动力学特性而言,
‘
s’对映异构体显著优于
‘
r’对映异构体。因此,在第二方面,本发明提供了由式ia表示的化合物(2s)-3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇:
[0034][0035]
和/或其药学上可接受的盐。
[0036]
因此,本发明的一个方面是一种组合物,其中存在于组合物中的式i的对映异构体的总量的至少75%是s对映异构体。在某些实施方案中,该百分比可以是,存在的式i的对映异构体的至少85%、至少90%、至少95%、至少99%和100%是s对映异构体。在其它实施方案中,所述组合物不含有式i的r对映异构体。
[0037]
在另一个方面,本发明提供了式ia的化合物或其药学上可接受的盐,其基本上不含有式ib的化合物
[0038][0039]
术语“基本上不含有式ib的化合物”表示,相对于式ia的化合物而言小于25%、小于20%、小于15%、小于10%、小于9%、小于8%、小于7%、小于6%、小于5%、小于4%、小于3%、小于2%、小于1%、小于0.9%、小于0.8%、小于0.7%、小于0.6%、小于0.5%、小于0.4%、小于0.3%、小于0.2%、小于0.1%、小于0.09%、小于0.05%或小于0.01%w/w的式ib的化合物的含量,或式ib的化合物不存在。
[0040]
因此,在一个实施方案中,本发明提供了式ia的化合物或其药学上可接受的盐,其中式ib的化合物的含量相对于式ia的化合物而言小于25%、小于20%、小于15%、小于
10%、小于5%、小于1%、小于0.5%、小于0.1%、小于0.05%、小于0.01%w/w或不存在。
[0041]
在另一个实施方案中,本发明提供了式ia的化合物或其药学上可接受的盐,其中式ia的化合物与式ib的化合物的对映异构体比大于75:25、大于80:20、大于85:15、大于90:10、大于95:5、大于96:4、大于97:3、大于98:2、大于99:1或为100:0。
[0042]
在另一个实施方案中,本发明提供了式ia的化合物或其药学上可接受的盐,其中式ia的化合物与式ib的化合物的对映异构体比大于80:20。在另一个实施方案中,式ia的化合物的对映异构体比大于85:15。在另一个实施方案中,式ia的化合物的对映异构体比大于90:10。在另一个实施方案中,式ia的化合物的对映异构体比大于95:5。在另一个实施方案中,式ia的化合物的对映异构体比大于96:4。在另一个实施方案中,式ia的化合物的对映异构体比大于97:3。在另一个实施方案中,式ia的化合物的对映异构体比大于98:2。在另一个实施方案中,式ia的化合物的对映异构体比大于99:1。在另一个实施方案中,式ia的化合物的对映异构体比是100:0,即式ib的化合物(r对映异构体)不存在。
[0043]
本发明还包括式i或式ia的化合物的前药或氘代衍生物。
[0044]
如本文中所述的本发明的化合物是特别具有er降解性能的er-拮抗剂,且因此被认为可用作药物,特别是用于治疗er依赖性的或er介导的疾病,诸如癌症,其选自、但不限于乳腺癌、卵巢癌、脑癌和子宫内膜癌。
[0045]
考虑到er-α在乳腺癌发展和进展中的核心作用,本发明的化合物可以单独地或与其它药剂联合地用于治疗乳腺癌,所述其它药剂包括、但不限于芳香酶抑制剂(诸如阿那曲唑、来曲唑等)、serm(诸如他莫昔芬、雷洛昔芬等)、抗雌激素剂(诸如氟维司群等)、黄体生成素-释放激素(lh-rh)激动剂(诸如亮丙瑞林等)、cdk4/6抑制剂(诸如帕博西尼等)或其它化学治疗剂,包括蒽环类、铂类、氮芥烷化剂等。
[0046]
因而,在另一个方面,本发明提供了在有此需要的人类中治疗er依赖性的或er介导的疾病或病症的方法,所述方法包括给其施用有效量的式i的化合物或其药学上可接受的盐。
[0047]
在另一个实施方案中,本发明提供了在有此需要的人类中治疗er依赖性的或er介导的疾病或病症的方法,所述方法包括给其施用有效量的式ia的化合物或其药学上可接受的盐。
[0048]
在另一个实施方案中,本发明提供了在有此需要的人类中治疗选自乳腺癌、子宫内膜癌、脑癌和卵巢癌的癌症的方法,所述方法包括给其施用有效量的式i的化合物或其药学上可接受的盐。
[0049]
在另一个实施方案中,本发明提供了在有此需要的人类中治疗选自乳腺癌、子宫内膜癌、脑癌和卵巢癌的癌症的方法,所述方法包括给其施用有效量的式ia的化合物或其药学上可接受的盐。
[0050]
在另一个实施方案中,本发明提供了治疗乳腺癌的方法,所述方法包括施用有效量的式i的化合物或式ia的化合物或其药学上可接受的盐。
[0051]
药物组合物
[0052]
可以将本文公开的化合物配制成组合物,该组合物另外包含合适的药学上可接受的载体,包括赋形剂和有助于将化合物施用给受试者的其它化合物。使用一种或多种药学上可接受的赋形剂,可以以常规方式配制本发明的药物组合物。药学上可接受的赋形剂可
以包括诸如稀释剂、崩解剂、粘合剂、润滑剂、助流剂、聚合物、包衣剂、溶剂、共溶剂、防腐剂、润湿剂、增稠剂、消泡剂、甜味剂、矫味剂、抗氧化剂、着色剂、增溶剂、塑化剂、分散剂、等物品。可以以丸剂、片剂、包衣片剂、胶囊剂、粉剂、颗粒剂、丸剂、贴剂、植入物、薄膜、液体、半固体、凝胶剂、气雾剂、乳剂、酏剂等的形式配制本发明的化合物。这样的药物组合物及其制备方法描述于例如remington:the science and practice of pharmacy(d.b.troy,编,第21版,lippincott,williams&wilkins,2006),其内容通过引用整体并入本文。在某些实施方案中,可以口服地、胃肠外地、肌肉内地、透皮地或静脉内地施用本文描述的化合物和组合物。
[0053]
因而,在一个实施方案中,本发明提供了一种药物组合物,其包含式i的化合物或式ia的化合物或其药学上可接受的盐以及药学上可接受的载体、稀释剂或赋形剂。
[0054]
用于治疗如本文所述的疾病的化合物的合适剂量可以由相关领域的技术人员确定。通常基于从动物研究衍生出的初步证据通过在人类中的剂量范围研究来鉴别治疗剂量。剂量必须足以产生期望的治疗益处,而不造成不希望的副作用。本领域技术人员也可以理解和调整施用模式、剂型和合适的药物赋形剂。
[0055]
参考下述实施例进一步详细说明本发明。应当理解,本文中的实施例仅仅是示例性的,且不限制本公开内容或所附权利要求的范围。
[0056]
制备方法
[0057]
如下所述制备式i的化合物、式ia的化合物和它们的密切相关的类似物。除非另外指出,否则所有的溶剂和试剂均按照从商业来源获得的状态使用。在氘代dmso中用在500mhz运行的波谱仪记录1h-nmr谱。
[0058]
通过本领域已知的方法可以将式i的化合物或式ia的化合物转化成它们的盐,所述方法包括、例如,将式i或式ia的化合物溶解在合适的溶剂中并用适当的酸处理它。
[0059]
实施例1:3-(3,5-二氟苯基)-2-{4-[(e)-3-(3-氟甲基氮杂环丁烷-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(式i的化合物)的制备
[0060][0061]
步骤i:2-(3,5-二氟苯基)-1-(2,4-二羟基苯基)乙酮的制备
[0062][0063]
在室温将草酰氯(5.98ml,0.070mol)逐滴加入3,5-二氟苯乙酸(10g,0.058mol)和n,n-二甲基甲酰胺(0.5ml)在二氯甲烷(100ml)中的搅拌溶液中并在室温搅拌30min。将反应混合物在减压下在30-35℃浓缩并然后溶解在二氯甲烷(20ml)中。在0-5℃将得到的溶液
加入间苯二酚(9.58g,0.087mol)和氯化铝(11.60g,0.087mol)在二氯甲烷(80ml)中的搅拌溶液中并在室温搅拌16小时。将反应物在0-5℃用2n盐酸溶液(120ml)缓慢地淬灭并在相同温度搅拌1小时。将固体过滤并接连地用水和正己烷洗涤。将得到的固体在真空下干燥以得到2-(3,5-二氟苯基)-1-(2,4-二羟基苯基)乙酮。
[0064]
步骤ii:2-(3,5-二氟苯基)-1-[2-羟基-4-(四氢吡喃-2-基氧基)苯基]乙酮的制备
[0065][0066]
在室温将3,4-二氢-2h-吡喃(45.58ml,0.50mol)加入2-(3,5-二氟苯基)-1-(2,4-二羟基苯基)乙酮(44.0g,0.167mol)和吡啶鎓对甲苯磺酸盐(6.28g,0.025mol)在二氯甲烷(880ml)中的搅拌溶液中。将反应混合物在室温搅拌2小时并用饱和碳酸氢钠水溶液淬灭。将有机层分离并将水层再次用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在减压下浓缩以得到粗制物质,将其在室温在正己烷:乙酸乙酯(95:5)的混合物中搅拌并过滤以得到2-(3,5-二氟苯基)-1-[2-羟基-4-(四氢吡喃-2-基氧基)-苯基]乙酮。
[0067]
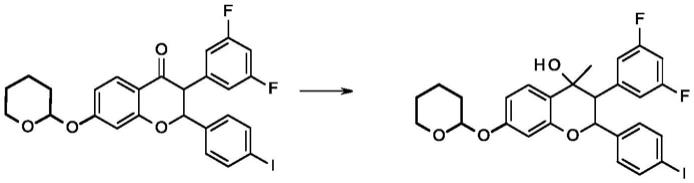
步骤iii:3-(3,5-二氟苯基)-2-(4-碘苯基)-7-(四氢吡喃-2-基氧基)色烷-4-酮的制备
[0068][0069]
将1,8-二氮杂双环(5.4.0)十一碳-7-烯(dbu,0.055g,0.00036mol)加入2-(3,5-二氟苯基)-1-[2-羟基-4-(四氢吡喃-2-基氧基)-苯基]乙酮(0.5g,0.0014mol)、4-碘苯甲醛(0.37g,0.0016mol)和哌啶(0.03g,0.00036mol)在异丙醇(10ml)中的搅拌浆中。将反应混合物加热至90-95℃保持3小时。在减压下除去溶剂,以得到3-(3,5-二氟苯基)-2-(4-碘苯基)-7-(四氢吡喃-2-基氧基)色烷-4-酮。
[0070]
步骤iv:3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-色烷-4-醇的制备
[0071][0072]
在20-25℃将在四氢呋喃(thf,1.6ml)中的甲基氯化镁(3m)加入3-(3,5-二氟苯基)-2-(4-碘苯基)-7-(四氢吡喃-2-基氧基)-色烷-4-酮(0.8g,0.0014mol)在无水thf(12ml)中的搅拌溶液中并搅拌1小时。将反应混合物在0-5℃用饱和氯化铵水溶液、随后用水淬灭。将有机层分离并将水层用乙酸乙酯再次萃取。将合并的有机层经无水硫酸钠干燥
并在真空下除去溶剂以得到标题化合物。
[0073]
步骤v:3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-2h-色烯-7-醇的制备
[0074][0075]
将浓硫酸(0.22ml,0.0042mol)在甲醇(2ml)中的溶液加入3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-色烷-4-醇(0.8g,0.0014mol)在甲醇(10ml)中的搅拌溶液中并在65-70℃加热3小时。将反应混合物冷却至0-5℃。将饱和碳酸氢钠溶液加入以上反应混合物并用乙酸乙酯萃取。将合并的有机层经无水硫酸钠干燥并在真空下除去溶剂以得到粗制物质,将其通过柱色谱法(硅胶,甲苯:乙酸乙酯(97:3))纯化以得到标题化合物。
[0076]
步骤vi:3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯的制备
[0077][0078]
在室温将3,4-二氢-2h-吡喃(10.34ml,0.113mol)加入3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-2h-色烯-7-醇(18g,0.038mol)和吡啶鎓对甲苯磺酸盐(1.42g,0.0057mol)在二氯甲烷(200ml)中的搅拌溶液中并搅拌16小时。将反应混合物用饱和碳酸氢钠溶液淬灭。将有机层分离并将水层用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在真空下除去溶剂以得到粗制物质,将其通过柱色谱法(硅胶,甲苯)纯化以得到标题化合物。
[0079]
步骤vii:(e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]苯基}丙烯酸乙酯的制备
[0080][0081]
在室温将丙烯酸乙酯(0.23g,0.0022mol)加入3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯(0.25g,0.00045mol)和三乙胺(0.37ml,0.0027mol)在n-甲基-2-吡咯烷酮(2ml)中的搅拌溶液中,随后加入pd(pph3)2cl2(0.003g,0.0000044mol)。将得到的反应混合物在95℃加热1小时。将水加入反应混合物并将产物用乙酸乙酯萃取。将合并的有机层经无水硫酸钠干燥并在真空下除去溶剂以得到粗制物质,
将其通过柱色谱法(硅胶,正己烷:乙酸乙酯(85:15))纯化以得到标题化合物。
[0082]
步骤viii:(e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}丙-2-烯-1-醇的制备
[0083][0084]
在-30℃将在甲苯(0.56ml,0.00079mol)中的二异丁基氢化铝(20%)溶液加入(e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}丙烯酸乙酯(0.14g,0.00026mol)在甲苯(5.6ml)中的搅拌溶液中并在-20至-25℃搅拌45min。在-20℃逐滴加入甲醇(0.5ml)和酒石酸钾钠(20%)溶液(5ml)。使反应混合物达到室温并在室温用水处理。将有机层分离并将水层用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在减压下除去溶剂以得到粗制物质,将其通过柱色谱法(硅胶,正己烷:乙酸乙酯(60:40))纯化以得到标题化合物。
[0085]
步骤ix:1-((e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}烯丙基)-3-氟甲基氮杂环丁烷
[0086][0087]
在0-5℃将碘(1.02g,0.0041mol)逐份加入三苯基膦(1.07g,0.0041mol)和咪唑(0.31g,0.0045mol)在二氯甲烷(10ml)中的搅拌溶液中。将反应混合物在0-5℃搅拌30min。在0-5℃将(e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]苯基}丙-2-烯-1-醇(1.0g,0.0020mol)在二氯甲烷(10ml)中的溶液加入该反应混合物并搅拌20分钟。将它缓慢地倒入冷碳酸氢钠溶液中,并用二氯甲烷萃取。将合并的有机层接连地用偏亚硫酸氢钠水溶液和盐水溶液洗涤。将有机层经无水硫酸钠干燥并在真空下除去溶剂以得到粗制物质。将正己烷:乙酸乙酯(9:1)的混合物(20ml)加入以上粗制物质并在室温搅拌30min。最后,将它过滤并将滤液在35-38℃在减压下浓缩以得到粗制物质,将其再次在正己烷:乙酸乙酯(9:1)的混合物(10ml)中搅拌30min。将它再次过滤并将滤液在35-38℃在减压下浓缩以得到3-(3,5-二氟苯基)-2-[4-((e)-3-碘丙烯基)苯基]-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯。
[0088]
将三乙胺(0.56ml,0.004mol)加入3-氟甲基氮杂环丁烷盐酸盐(0.38g,0.003mol)在乙腈(10ml)中的搅拌溶液中。将反应混合物在室温搅拌30分钟。在室温将3-(3,5-二氟苯基)-2-[4-((e)-3-碘丙烯基)苯基]-4-甲基-7-(四氢-吡喃-2-基氧基)-2h-色烯(1.2g,0.002mol)在乙腈(10ml)中的溶液加入反应混合物并继续搅拌45分钟。将水加入反应混合物并用乙酸乙酯萃取。将有机层分离,经无水硫酸钠干燥并在减压下除去溶剂以得到粗制
物质,将其通过柱色谱法(硅胶,二氯甲烷:甲醇(97:3))纯化以得到标题化合物。
[0089]
步骤x:3-(3,5-二氟苯基)-2-{4-[(e)-3-(3-氟甲基氮杂环丁烷-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(式i)的制备
[0090][0091]
在0-5℃将硫酸(0.75ml,0.014mol)在甲醇(70ml)中的溶液加入1-((e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}烯丙基)-3-氟甲基氮杂环丁烷(7.6g,0.014mol)在甲醇(20ml)中的搅拌溶液中。将反应混合物在室温搅拌30分钟。在0-5℃加入饱和碳酸氢钠溶液和水并用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在减压下浓缩以得到残余物,将其通过柱色谱法(硅胶,二氯甲烷:甲醇(90:10))纯化以得到标题化合物。
[0092]
实施例2:(2s)-3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇(式ia的化合物)和(2r)-3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇(式ib的化合物)的制备
[0093][0094]
将来自实施例1的外消旋混合物的对映异构体通过手性hplc(柱:od-h(250x30mm,5μ);流动相-正己烷:乙醇:二乙胺900:100:1)分离,其中r对映异构体(式ib的化合物)首先洗脱,其次是期望的s对映异构体(式ia的化合物)。进一步,使用以下试验条件确定式ia的化合物的比旋光度(sor):
[0095]
浓度:1%w/v在丙酮中;
[0096]
温度:25℃;
[0097]
光源:钠灯(d线);
[0098]
式ia的化合物的sor:
[0099]
按照以下分析条件通过hplc确定式ia的化合物的手性纯度:
[0100]
柱:od-3(250x 4.6)mm 3μm
[0101]
流动相:正己烷/乙醇/二乙胺(90/10/0.1,v/v/v)
[0102]
流速:1.0ml/min;柱温度:25℃;检测器:uv:230nm;
[0103]
样品浓度:0.5mg/ml
[0104]
稀释剂:流动相。
[0105]
式ia的化合物的手性纯度=99.69:0.31(s:r);相对于式ib的化合物的相对保留时间(rrt)=约1.1
[0106]
实施例3:3-(3,5-二氟苯基)-2-[4-[(e)-3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-6-醇(化合物2)的制备
[0107][0108]
按照实施例1(步骤iii-步骤x)的类似方法制备外消旋的化合物2,其中在步骤iii中使用2-(3,5-二氟苯基)-1-(2-羟基-5-四氢吡喃-2-基氧基-苯基)乙酮替代2-(3,5-二氟苯基)-1-[2-羟基-4-(四氢吡喃-2-基氧基)-苯基]乙酮。
[0109]
实施例4:3-(3,5-二氟苯基)-2-{4-[(e)-3-(4-氟甲基哌啶-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(化合物3)的制备
[0110][0111]
步骤i:1-((e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}烯丙基)-4-氟甲基哌啶
[0112][0113]
在0-5℃将甲磺酰氯(0.11ml,1.4mmol)在二氯甲烷(1ml)中的溶液逐滴加入(e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]苯基}丙-2-烯-1-醇(0.57g,1.17mmol)和三乙胺(0.24ml,1.75mmol)在二氯甲烷(5ml)中的搅拌溶液中。将反应混合物在0-5℃进一步搅拌30分钟。将水加入反应混合物并将有机层分离。将水层用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并用于下一步。在0-5℃将该溶液加入三乙胺(0.65ml,4.7mmol)和4-氟甲基哌啶盐酸盐(0.54g,3.5mmol)在乙腈(6ml)中的溶液中。将反应混合物在室温搅拌1.5小时。加入水并将混合物用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在减压下浓缩以得到粗制物质,将其通过柱色谱法(硅胶,甲醇:二氯甲烷(5:95))纯化以得到标题化合物。
[0114]
步骤ii:3-(3,5-二氟苯基)-2-{4-[(e)-3-(4-氟甲基哌啶-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(化合物3)
[0115][0116]
将1-((e)-3-{4-[3-(3,5-二氟苯基)-4-甲基-7-(四氢吡喃-2-基氧基)-2h-色烯-2-基]-苯基}烯丙基)-4-氟甲基哌啶(0.7g,1.18mmol)在硫酸(0.07ml)和甲醇(5ml)的混合物中的溶液在室温搅拌10分钟。将反应混合物用饱和碳酸氢钠溶液呈碱性,并用二氯甲烷萃取。将合并的有机层经无水硫酸钠干燥并在减压下浓缩以得到粗制物质,将其通过柱色谱法(硅胶,甲醇:二氯甲烷(8:92))纯化以得到标题化合物。
[0117]
实施例5:3-(3,5-二氟苯基)-2-[4-[(e)-3-[(3r)-3-(氟甲基)吡咯烷-1-基]丙-1-烯基]苯基]-4-甲基-2h-色烯-7-醇(化合物4)的制备
[0118][0119]
按照实施例4的类似方法,在步骤i中使用(3r)-3-(氟甲基)吡咯烷盐酸盐替代4-氟甲基哌啶盐酸盐,制备化合物4。
[0120]
实施例6:3-(3,5-二氟苯基)-4-甲基-2-[4-[(e)-3-[(3r)-3-甲基吡咯烷-1-基]丙-1-烯基]苯基]-2h-色烯-7-醇(化合物5)的制备
[0121][0122]
按照实施例4的类似方法,在步骤i中使用(3r)-3-甲基吡咯烷盐酸盐替代4-氟甲基哌啶盐酸盐,制备化合物5。
[0123]
实施例7:3-(3,5-二氟苯基)-2-{4-[(z)-3-(3-氟甲基氮杂环丁烷-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(化合物6)的制备
[0124][0125]
步骤i:3-[4-[3-(3,5-二氟苯基)-4-甲基-7-四氢吡喃-2-基氧基-2h-色烯-2-基]苯基]丙-2-炔-1-醇
[0126][0127]
将双(三苯基膦)二氯化钯(ii)(0.125g,0.18mmol)加入3-(3,5-二氟苯基)-2-(4-碘苯基)-4-甲基-7-四氢吡喃-2-基氧基-2h-色烯(2.0g,3.6mmol)、丙炔醇(0.60g,10.7mmol)和碘化亚铜(i)(0.054g,0.29mmol)在四氢呋喃:三乙胺的混合物(1:1,64ml)中的搅拌溶液中。在室温继续搅拌2小时。将反应混合物在减压下浓缩以得到粗残余物,将其通过柱色谱法(硅胶,甲苯-乙酸乙酯(4:1))纯化以得到标题化合物。
[0128]
步骤ii:1-[3-[4-[3-(3,5-二氟苯基)-4-甲基-7-四氢吡喃-2-基氧基-2h-色烯-2-基]苯基]丙-2-炔基]-3-(氟甲基)氮杂环丁烷
[0129][0130]
按照实施例4、步骤i的类似方法制备步骤ii化合物。
[0131]
步骤iii:1-[(z)-3-[4-[3-(3,5-二氟苯基)-4-甲基-7-四氢吡喃-2-基氧基-2h-色烯-2-基]苯基]烯丙基]-3-(氟甲基)氮杂环丁烷
[0132]
[0133]
将lindlar催化剂(0.125g,25%w/w)加入1-[3-[4-[3-(3,5-二氟苯基)-4-甲基-7-四氢吡喃-2-基氧基-2h-色烯-2-基]苯基]丙-2-炔基]-3-(氟甲基)氮杂环丁烷(0.50g,0.89mmol)和喹啉(0.05g,10%w/w)在乙醇(10ml)中的溶液中。将反应混合物使用氢气填充的气舱在氢气氛下在室温搅拌5小时。将反应混合物过滤,并用乙醇洗涤。将滤液在减压下浓缩以得到粗制物质,将其通过柱色谱法(硅胶,二氯甲烷:甲醇(97:3))纯化以得到标题化合物。
[0134]
步骤iv:3-(3,5-二氟苯基)-2-{4-[(z)-3-(3-氟甲基氮杂环丁烷-1-基)丙烯基]苯基}-4-甲基-2h-色烯-7-醇(化合物6)
[0135][0136]
使用实施例4(步骤ii)的类似方法除去thp保护以得到外消旋的化合物6。
[0137]
实施例8:3-(3,5-二氟苯基)-2-[4-[3-[3-(氟甲基)氮杂环丁烷-1-基]丙-1-炔基]苯基]-4-甲基-2h-色烯-7-醇(化合物7)的制备
[0138][0139]
使用实施例4、步骤ii的类似方法,通过除去实施例7(步骤ii)化合物的thp保护,制备外消旋的化合物7。
[0140]
下面提供了式i、式ia的化合物和有关类似物的1h nmr数据:
[0141]
[0142]
[0143]
[0144][0145]
#
在2h-色烯环的第2个位置的手性。
[0146]
通过下述测定确定本公开内容的化合物的生物活性:
[0147]
选择性雌激素受体降解(serd)测定:
[0148]
在携带er野生型的mcf-7细胞和携带突变体er(wt/d538g和wt/y537s)的mcf-7细胞中评价了试验化合物的serd活性。简而言之,将细胞铺板在补充有5%木炭剥离的胎牛血清的无酚红rpmi1640培养基中。在48孔板中,mcf-7wt和mcf-7d538g的接种密度为40000个细胞/孔,mcf-7y537s的接种密度为50000个细胞/孔。在温育过夜后,将细胞用不同浓度的试验化合物(最终浓度:1000nm至0.01nm,0.1%dmso)处理4天。使用补充有1mm edta、0.5%triton x-100、5mm naf、6m尿素和1x蛋白酶抑制剂混合液的pbs裂解细胞。使用蛋白质印迹分析裂解物中的erα蛋白。对于蛋白质印迹,将细胞裂解物(12.5-40μg总蛋白)在10%sds page上分离并转移到pvdf膜上。将印迹使用在0.1%pbs-t中的5%脱脂奶粉在室温封闭1小时,然后与兔抗-人β-肌动蛋白抗体和兔抗-人erα抗体在室温共温育2小时。随后将印迹用抗-兔igg-hrp缀合物作为第二抗体在室温下探测1小时。将印迹使用west pico super signal化学发光底物显色,并处理条带用于使用image lab软件(biorad 6.0.0版)进行密度测定法分析。将erα条带强度标准化至各个样品的持家蛋白。通过相对于媒介物对照(作为100%)标准化来计算er剩余百分比。
[0149]
mcf-7细胞生长抑制测定:
[0150]
在生长抑制测定中评价试验化合物的抗增殖活性。简而言之,将携带er野生型(wt)的mcf-7细胞和携带突变体er(wt/d538g和wt/y537s)的mcf-7细胞以1000个细胞/孔的密度接种在96-孔板内的补充有10%木炭剥离的胎牛血清的无酚红rpmi1640培养基中。将细胞在37℃和5%co2下温育过夜,然后加入在dmso中的不同浓度的试验化合物。孔中dmso的最终浓度为0.5%。将细胞与试验化合物一起温育七天后,使用prestoblue
tm
试剂评估细胞生存力。通过使用媒介物对照作为0%增殖抑制将细胞生存力标准化,计算细胞增殖的抑制百分比。
[0151]
在下面表1中显示了1nm浓度的式i的化合物和它的密切相关的化合物的er-α降解测定的结果。
[0152]
表1:er-α降解测定
[0153][0154]
从上表显而易见,与包括其位置异构体化合物2在内的在结构上接近的化合物相比,式i的化合物在mcf-7细胞系(野生型)中显示出显著更多的er-α降解。令人惊讶地发现,如在化合物2中,将式i的化合物中羟基的位置从色烯部分的第7位变为第6位,导致serd活性的完全丧失。在侧链中具有6-或5-元杂环烷基环的化合物3、4和5具有可忽略的serd活性(超过80%的er剩余),而在侧链中具有4-元氮杂环丁烷环的式i的化合物表现出良好的er降解(仅约41%的er剩余)。还令人惊讶地发现,如在化合物7中,将式i的化合物的侧链中的双键改变为三键,导致serd活性的完全丧失。此外,作为式i的化合物的几何异构体(顺式异构体)的化合物6显示出非常微不足道的serd活性。
[0155]
因此,式i的化合物具有有效的选择性雌激素受体降解剂所需的所有最佳结构属性。本发明的发明人进一步将式i的化合物拆分为其s和r立体异构体。在本公开内容中,将s异构体表示为式ia,而将r异构体表示为式ib。
[0156][0157]
进一步测试了式ia的化合物和式ib的化合物在生长抑制测定(mcf-7细胞系)中的抗增殖活性和er-α降解。提供的结果如下表2所示。
[0158]
表2:式ia的化合物与其r对映异构体即式ib的化合物的体外效能的对比
[0159][0160]
如上表所证明的,式ia的化合物在mcf-7生长抑制测定(野生型)中表现出优于式ib的化合物的多倍优异活性,其中式ia的化合物的效力比式ib的化合物强约233倍。式ia的化合物在突变细胞系(y537s和d538g)中也显示出类似的趋势。还发现式ia的化合物在er-α降解测定中比其r-对映异构体的效力强多倍,其中式ia的化合物在er-α降解(野生型)中比式ib的化合物的效力强约34倍。r对映异构体在突变细胞系(y537s和d538g)中没有显示出er-α降解。
[0161]
药代动力学研究:
[0162]
在药物发现中,为了成为用于口服施用的良好药物候选物,具有化合物的良好口服生物利用度和药代动力学(pk)特性是非常重要的。许多具有有效体外效力的化合物未能在人体中显示出治疗效果。这些化合物的治疗效果的缺乏主要是由于它们不适当的药代动力学性能,诸如低生物利用度、短半衰期、快速代谢或快速清除,从而导致短的作用持续时间。因此,良好的药物候选物不仅应具有良好的效能,还应具有良好的药代动力学特性。按照以下提供的程序确定式ia和ib的化合物的药代动力学特性。
[0163]
在单次口服剂量施用后,在雌性sd大鼠(n=2)中评价pk曲线。将大鼠在施用前禁食12-13小时,并在口服治疗后2小时提供饲料。在0.5%羧甲基纤维素(cmc)中的吐温80(悬浮液总体积的0.4%v/v)中制备化合物的剂量制剂。以50mg/kg口服施用治疗。为了评价血浆浓度,在治疗后0.25、1、4、8、24、48和72小时时间点,在含有肝素钠作为抗凝剂(100iu/ml)的埃彭道夫管中,通过眶后网状组织从大鼠抽取0.2ml血液,并立即在4℃在8500rpm离心7分钟。将血浆分离并在-70℃储存直到分析。按照以下给出的方法进行样品的分析:
[0164]
工作校准标准品和质量控制标准品制备:
[0165]
制备具有0.10、0.20、2.00、4.00、8.00、12.00、16.00和20.00μg/ml浓度的工作校准标准品。在0.30、3.00、10.00、15.00和60.00μg/ml的浓度制备质量控制标准品。
[0166]
工作内部标准品(wis)的制备:
[0167]
将准确地称量的5mg卡马西平转移进100ml容量瓶中,用稀释剂(0.1%甲酸水溶液:乙腈30:70v/v)溶解并稀释至刻度。由上述溶液制备5μg/ml的wis。
[0168]
样品处理:
[0169]
将5μl工作标准品和质量控制品掺入到95μl空白血浆中。将来自以上掺杂的标准品和质量控制品的25μl等分到微量离心管中。还将25μl研究样品等分到微量离心管中。将5μl wis以零标准品加入到线性和研究样品(空白除外)中。将样品进一步涡旋10-15秒。将
150μl milli q水加入所有制备的样品中并涡旋10-15秒。将样品加载到预处理的筒上(用1ml甲醇、再用1ml milli q水进行预处理)。用2x 1ml milli q水洗涤筒。用hplc管形瓶中的1ml洗脱溶液{乙腈:0.1%甲酸溶液(70:30)}进行洗脱。
[0170]
lc-ms/ms方法:
[0171]
用分流器用1.25ml/min的流速和20μl的注射体积在chiral pack id(250*4.6mm,5μ)上进行色谱分离。将样品冷却器维持在10℃。将柱恒温箱温度设置为30℃。流动相由以下组成:
[0172]
流动相a:将1.47g碳酸氢铵加入1l水、1ml dea,ph 9.2
±
0.1
[0173]
流动相b:甲醇,
[0174]
其中流动相a与流动相b分别以30:70v/v的比例混合。式ia的化合物、式ib的化合物和内部标准品的保留时间分别为约8.7、7.6和4.2min。总色谱运行时间为16分钟。
[0175]
通过串联质谱法(tsq quantum,discovery max,thermo electron corporation)进行检测,并使用lcquan软件2.9版qf1对峰面积进行积分。将检测器设置在mrm模式,其中对于式ia的化合物或式ib的化合物监测477.900m/z
→
272.982m/z(ce 20)的跃迁,且对于内部标准品监测237.058m/z
→
194.003m/z(ce 16)的跃迁。
[0176]
药代动力学研究的结果如下表3所示。
[0177]
表3:式i和式i的化合物的pk研究
[0178][0179]
从上表显而易见,式ia的化合物与其r-对映异构体相比表现出优异的药代动力学特性。发现式ia的化合物的c
max
比式ib的化合物的c
max
大了约2.9倍。类似地,发现式ia的化合物的auc
last
比式ib的化合物的auc
last
大了约4.2倍。
[0180]
式i的化合物和式ia的化合物的体内效力:
[0181]
在携带皮下mcf7-y537s异种移植物的雌性无胸腺裸鼠中评价了式i的化合物和式ia的化合物的效力。在0.5%w/v羧甲纤维素(cmc)中的吐温80(悬浮液总体积的0.4%v/v)中制备式i的化合物和式ia的化合物的剂量制剂,并每天口服施用一次,持续28天。用数字游标卡尺测量肿瘤的两个垂直直径。使用下述方程式计算肿瘤体积(v):v=(a
2 x b)/2,其中“a”是肿瘤的宽度(小直径),且“b”是长度(大直径),均以毫米为单位。每周两次监测肿瘤并与媒介物治疗组进行对比。研究的结果如下表4所示。与媒介物组相比,式ia和式i的化合物均显示出肿瘤生长的显著降低(参见图1)。结果表明,与媒介物组相比,50mg/kg的式ia的化合物和在100mg/kg剂量水平的式i的外消旋化合物显示出相似的肿瘤生长抑制(tgi),分别具有56%和57%tgi。
[0182]
表4:式ia的化合物和式i的化合物在mcf7-y537s小鼠异种移植物中的体内效力
[0183][0184]
总之,与它们的密切相关的化合物相比,本公开内容的化合物在体外测定中(例如在er-α降解测定中)显示出更好的效能。本公开内容的化合物还在mcf7-y537s异种移植物中也显示出良好的体内效力。本公开内容的化合物、特别是式ia的化合物显示出良好的药代动力学特性,且因此可以适合口服施用。本公开内容的化合物或其药学上可接受的盐可以配制成口服剂型并且可以用于治疗与er的调节相关的疾病,诸如er-阳性的乳腺癌。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1