新化合物和方法与流程
1.本发明涉及作为c-abl之抑制剂的式(i)化合物。本发明还涉及包含这些化合物的药物组合物,以及其在治疗或预防其中c-abl的抑制是有益的医学病症中的用途。这样的医学病症包括神经退行性疾病和癌症。
背景技术:
2.abl1(艾贝尔森鼠白血病病毒癌基因同源物1(abelson murine leukaemia viral oncogene homolog 1))是显示酪氨酸激酶酶活性并与多种细胞功能相关的蛋白质。在人中,该蛋白质由位于染色体9上的abl1基因编码。在哺乳动物基因组内发现的abl1基因的形式表示为c-abl。
3.费城染色体(philadelphia chromosome)是由t(9,22)染色体相互易位形成的染色体22中的遗传异常,导致称为bcr-abl1的融合基因。该融合基因包含来自染色体9的abl1基因和bcr基因的一部分。abl1蛋白的酪氨酸激酶活性通常受到严格调节,然而,融合基因中的bcr结构域导致abl1激酶的组成型激活。然而,bcr-abl和c-abl的结合结构域是相同的。
4.c-abl的激活涉及多种疾病,特别是癌症。例如,bcr-abl突变的存在与慢性髓性白血病(chronic myeloid leukaemia,cml)强相关。其也在急性淋巴母细胞白血病(acute lymphoblastic leukaemia,all)的一些情况中被发现。尼洛替尼(nilotinib)和帕纳替尼(ponatinib)二者均为结合在c-abl的atp位点中并且已用于治疗慢性髓性白血病(cml)和急性淋巴母细胞白血病(all)的正构c-abl抑制剂。
5.asciminib(abl001)是结合在c-abl的肉豆蔻酸酯口袋中的变构c-abl抑制剂。ascminib目前正处于治疗cml和费城染色体阳性all的临床试验中,其作为独立治疗或者与c-abl的正构酪氨酸激酶抑制剂(例如尼洛替尼、帕纳替尼、达沙替尼(dasatinib)和博舒替尼(bosutinib))结合。
6.可通过c-abl抑制治疗的白血病的范围包括慢性髓性白血病(cml)、急性淋巴母细胞白血病(all)、急性髓性白血病(acute myelogenous leukaemia,aml)、混合表型急性白血病(mixed-phenotype acute leukaemia,mpal)及其中枢神经系统(central nervous system,cns)转移。
7.c-abl的激活也涉及神经退行性疾病。神经退行性疾病可被神经元的进行性变性和最终死亡表征。具体的神经退行性疾病包括肌萎缩侧索硬化(amyotrophic lateral sclerosis,als)和帕金森病(parkinson’s disease,pd)。
8.als是由运动神经元的进行性变性引起的致命的神经退行性疾病。据报道,c-abl信号传导激活有助于神经元凋亡,并且c-abl抑制剂可防止运动神经元死亡[rojas et al.frontiers in cellular neuroscience,2015,9,203;imamura et al.science translational medicine,2017]。
[0009]
帕金森病(pd)是由黑质致密部(substantia nigra pars compacta)中多巴胺能
novel antifibrotic agent in bleomycin-induced pulmonary fibrosis in mice am.j.respir.crit.care med.2005,171,1279-85)。另一项研究表明伊马替尼(imatinib)和尼洛替尼二者均减弱了小鼠中经博来霉素诱导的急性肺损伤和肺纤维化(rhee et al.effect of nilotinib on bleomycin-induced acute lung injury and pulmonary fibrosis in mice.respiration 2011,82,273-87)。尽管在这些研究中,作者聚焦于与pdgfr有关的机制的意义,但有趣的是rhee et al.(respiration.2011,82,273-87)的研究,比伊马替尼更强效的c-abl抑制剂尼洛替尼显示出优异的治疗性抗纤维化作用,因此支持c-abl抑制剂用于治疗具有肺部炎症的人疾病的治疗适用性。在另一项研究中,将小鼠暴露于高氧提高了发动蛋白2磷酸化和活性氧物质产生和肺渗漏所需的abl1激活(singleton et al.dynamin 2 and c-abl are novel regulators of hyperoxia-mediated nadph oxidase activation and reactive oxygen species production in caveolin-enriched microdomains of the endothelium j.biol.chem.2009,284,34964-75)。
[0016]
也有报道称,c-abl抑制可能是用于缓解重症急性胰腺炎中的局部和系统炎症的有用策略[r madhi et al,journal of leukocyte biology,2019,106(2):455-466]。此外,bcr-abl抑制剂已用于治疗肺动脉高压[d.dumitrescu et al,european respiratory journal,2011,38:218-220]。
[0017]
鉴于以上,对可用于治疗和预防其中c-abl的抑制是有益的之医学病症,例如神经退行性疾病(即als和pd)和癌症(尤其是白血病)的新化合物的需求尚未满足。
技术实现要素:
[0018]
出乎意料地,已发现式(i)化合物抑制c-abl并且因此治疗或预防上述医学病症。不希望受理论束缚,认为式(i)化合物结合在c-abl的肉豆蔻酸酯袋中并且因此通过变构抑制机制起作用。
[0019]
此外,与已知化合物相比,式(i)化合物具有导致用作药物的潜力提高的某些有益的特性。这可依据其效力、脑与血浆比值、生物利用度、清除率、半衰期、溶解度、选择性分布(例如激酶选择性)、低herg抑制活性和/或其它显著的药代动力学特性。
[0020]
因此,本发明涉及式(i)化合物,
[0021][0022]
或其可药用盐、溶剂合物、水合物、几何异构体、互变异构体、光学异构体、n-氧化物和/或其前药,其中
[0023]
r1选自h和卤素;
[0024]
r2选自-ocf2cl、-ocf3、-scf3、-scf2cl、-cf2cf3、-cf2cf2cl、-ocf2cf3、-sf5、of2ch3、-socf3、-so2cf3、-ocf2cf2h和-scf2h;
[0025]
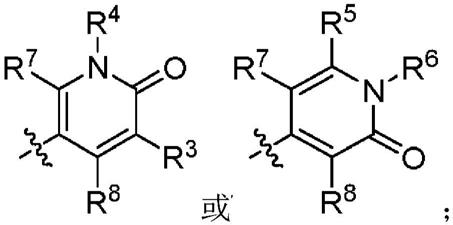
a为
[0026]
r3、r4、r5和r6独立地选自:
[0027]
(i)h、卤素、-oh、-c(o)nrdre、-nrarb、氰基、-c(o)orc和-c(o)rc;
[0028]
(ii)c
1-c7烷基、c
2-c6烯基、c
2-c6炔基、c
1-c6烷氧基,任选地经一个或更多个独立地选自-nrarb、氰基、-orc、卤素、氧代、6至10元芳基、5至10元杂芳基和4至10元杂环的取代基取代,任选地其中所述5至10元杂芳基和4至10元杂环独立地经一个或更多个选自卤素和c
1-c7烷基的取代基取代,其中所述c
1-c7烷基任选地经一个或更多个卤素原子取代;
[0029]
(iii)6至10元芳基和5至10元杂芳基,任选地经一个或更多个独立地选自卤素、-c(o)nrdre、-nrdre、-oh、氧代、氰基、-c(o)orc、和-c(o)rc、c
1-c7烷基、c
2-c6烯基、c
2-c6炔基、c
1-c6烷氧基、(c
1-c3烷基)-o-(c
1-c3烷基)和4至10元杂环的取代基取代,其中所述4至10元杂环任选地经氧代基团取代,其中所述烷基、烯基和炔基各自任选地独立地经一个或更多个卤素原子取代;和
[0030]
(iv)4至10元杂环,任选地经一个或更多个独立地选自卤素、氧代、-c(o)nrdre、-nrdre、氰基、-c(o)orc、和-c(o)rc、c
1-c7烷基、c
2-c6烯基、c
2-c6炔基、c
1-c6烷氧基、(c
1-c3烷基)-o-(c
1-c3烷基)、6至10元芳基、5至10元杂芳基和4至10元杂环的取代基取代,其中所述烷基、烯基和炔基各自任选地独立地经一个或更多个卤素原子取代;
[0031]
r7和r8各自独立地选自h和卤素;
[0032]
ra、rb和rc各自独立地选自h和c
1-c7烷基,其中所述c
1-c7烷基任选地经一个或更多个卤素原子取代;以及
[0033]
rd和re各自独立地选自h和c
1-c7烷基,其中所述c
1-c7烷基任选地经一个或更多个卤素原子取代,或者rd和re可以与其所连接的氮原子一起形成5或6元饱和、部分饱和或不饱和的环,其中所述环包含一个或更多个杂原子。
[0034]
这些化合物为本发明的化合物。
[0035]
优选的是,r1选自h、f和cl,并且更优选地r1为h。
[0036]
r7和r8各自还优选地选自h、f和cl,更优选h和f,并且最优选h。
[0037]
在本发明的一个优选特征中,r1以及各个r7和r8为h。
[0038]
在本发明的另一个优选特征中,r2选自-ocf2cl和-ocf3。
[0039]
尽管有以上所述,但是r4和r6上与分子其余部分的连接点不是卤素原子,并且优选不是杂原子,除了在形成n-氧化物的情况下。因此,r4和r6不是卤素,并且r4和r6优选不是-nrarb或c
1-c6烷氧基。
[0040]
在基团a上相对于酰胺接头的间位(即在位置r3、r4或r5,如式(i)中存在)包含芳族或杂芳族取代基可以是特别有利的,因为这可能导致提高的对c-abl的抑制。因此,特别优选的式(i)化合物为其中r5和r3和/或r4选自以上列出的取代基(iii)的那些。尤其优选的是,r5或r4选自取代基(iii)。
[0041]
高度优选的是,r1、r7和r8为h并且r2选自-ocf2cl和-ocf3。在这种情况下,本发明的化合物是式(ii)化合物,
[0042][0043]
其中x为f或cl;
[0044]
其中a为以及
[0045]
r3、r4、r5和r6如上限定。
[0046]
其中x为cl的式(ii)化合物是特别优选的,因为这些化合物可以表现出提高的对c-abl的抑制。
[0047]
在本发明的一个优选特征中,r3、r4、r5和r6独立地选自:
[0048]
(i)h和氰基;
[0049]
(ii)c
1-c6烷基和c
1-c6烷氧基,任选地经一个或更多个独立地选自卤素和5或6元杂环的取代基取代;
[0050]
(iii)5或6元杂环,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;和
[0051]
(iv)苯基,任选地经一个或更多个独立地选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基和4元杂环的取代基取代,其中所述4元杂环任选地经氧代基团取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0052]
(v)5至10元杂芳基,任选地经一个或更多个独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基和(c
1-c3烷基)-o-(c
1-c3烷基)的取代基取代,其中所述烷基任选地经一个或更多个卤素原子取代。
[0053]
在本发明的该优选特征中,特别优选的式(ii)化合物为其中r5和r3和/或r4选自取代基(iv)和(v)的那些。尤其优选的是,r5或r4选自取代基(iv)和(v)。
[0054]
在本发明的一个方面中,式(ii)化合物为式(iia)化合物,
[0055][0056]
其中r3和r4独立地选自:
[0057]
(i)h和氰基;
[0058]
(ii)c
1-c6烷基和c
1-c6烷氧基,任选地经一个或更多个独立地选自卤素和5或6元杂环的取代基取代;
[0059]
(iii)5或6元杂环,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;和
[0060]
(iv)苯基,任选地经一个或更多个独立地选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基和4元杂环的取代基取代,其中所述4元杂环任选地经氧代基团取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0061]
(v)5至10元杂芳基,任选地经一个或更多个独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基和(c
1-c3烷基)-o-(c
1-c3烷基)的取代基取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0062]
特别优选的式(iia)化合物为其中r3和/或r4选自取代基(iv)和(v)的那些。尤其优选的是,r4选自取代基(iv)和(v)。
[0063]
优选的是,r3选自:
[0064]
(i)h、c
1-c6烷氧基和氰基;
[0065]
(ii)c
1-c6烷基,任选地经一个或更多个卤素原子取代;
[0066]
(iii)苯基和5或6元杂芳基,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;和
[0067]
(iv)5或6元杂环,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代。
[0068]
特别优选的式(iia)化合物为其中r3选自取代基(iii)(即任选地经取代的苯基和5或6元杂芳基)的那些。
[0069]
作为r3的特别优选的5或6元杂芳基的实例包括:
[0070][0071]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。更优选地,它们包括:
[0072][0073]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。
[0074]
此外,优选的是,r4选自:
[0075]
(i)h:
[0076]
(ii)c
1-c6烷基,其中所述c
1-c6烷基任选地经一个或更多个选自卤素和5或6元杂环的取代基取代;
[0077]
(iii)苯基,任选地经一个或更多个独立地选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基和4元杂环的取代基取代,其中所述4元杂环任选地经氧代基团取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;
[0078]
(iv)5或6元杂芳基,任选地经一个或更多个独立地选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基和(c
1-c3烷基)-o-(c
1-c3烷基)的取代基取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0079]
(v)5或6元杂环,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;
[0080]
(vi)基团b
[0081][0082]
其中y和z各自独立地选自c、s、o和n,
[0083]
至少一个y或z为s、o或n,
[0084]
y和z各自任选地独立地经卤素或c
1-c6烷基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代,
[0085]
n为0或1,
[0086]
m为0或1;
[0087]
(vii)基团c
[0088][0089]
其中y和z各自独立地选自c、s、o和n,
[0090]
至少一个y或z为s、o或n,
[0091]
y和z各自任选地独立地经卤素或c
1-c6烷基取代,其中所述c
1-c6烷基任选地经一
个或更多个卤素原子取代,
[0092]
n为0或1,
[0093]
m为0或1;和
[0094]
(viii)基团d
[0095][0096]
其中y和z各自独立地选自c、s、o和n,
[0097]
至少一个z为c,
[0098]
y和z各自任选地独立地经卤素或c
1-c6烷基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代,
[0099]
r9选自卤素或c
1-c6烷基,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代,
[0100]
n为0或1;
[0101]
特别优选的式(iia)化合物为其中r4选自取代基(iii)、(iv)、(vi)、(vii)和(viii)的那些。
[0102]
作为r4的示例性5或6元杂芳基包括:
[0103][0104]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。更优选地,它们包括:
[0105][0106]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。
[0107]
作为r4的基团b、c和d的特别优选的实例包括:
[0108][0109]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。
[0110]
在本发明的另一个方面中,式(ii)化合物为式(iib)化合物,
[0111][0112]
其中r5和r6独立地选自:
[0113]
(i)h和氰基;
[0114]
(ii)c
1-c6烷基和c
1-c6烷氧基,任选地经一个或更多个独立地选自卤素和5或6元杂环的取代基取代;
[0115]
(iii)5或6元杂环,任选地经一个或更多个独立地选自卤素和c
1-c6烷基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代;和
[0116]
(iv)苯基,任选地经一个或更多个独立地选自卤素、氰基、c
1-c6烷基、c
1-c6烷氧基和4元杂环的取代基取代,其中所述4元杂环任选地经氧代基团取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0117]
(v)5至10元杂芳基,任选地经一个或更多个独立地选自卤素、氰基、氧代、c
1-c6烷基、c
1-c6烷氧基和(c
1-c3烷基)-o-(c
1-c3烷基)的取代基取代,其中所述烷基任选地经一个或更多个卤素原子取代;
[0118]
特别优选的式(iib)化合物为其中r5选自取代基(iv)和(v)的那些。
[0119]
优选的是,r5为5或6元杂芳基,任选地经一个或更多个独立地选自卤素和c
1-c6烷
基的取代基取代,其中所述c
1-c6烷基任选地经一个或更多个卤素原子取代。
[0120]
作为r5的示例性5或6元杂芳基包括:
[0121][0122]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。更优选地,它们包括:
[0123][0124]
或其互变异构体,其各自可以是任选地经取代的,如上文所概述。
[0125]
此外,优选的是,r6选自h和c
1-c6烷基,其中所述c
1-c6烷基任选经一个或更多个卤素原子取代。
[0126]
本发明的特定化合物是以下列出的那些。
[0127]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(3-乙氧基-1-甲基-吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0128]
·
1-(2-吗啉代乙基)-6-氧代-n-[4-(三氟甲氧基)苯基]吡啶-3-甲酰胺;
[0129]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1-甲基-1h-吡唑-3-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0130]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-[(3s)-四氢呋喃-3-基]吡啶-3-甲酰胺;
[0131]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-(1h-吡唑-5-基)吡啶-3-甲酰胺;
[0132]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-[(3r)-四氢呋喃-3-基]吡啶-3-甲酰胺;
[0133]
·
n-[4-(氯二氟甲氧基)苯基]-1-[1-(二氟甲基)-1h-吡唑-3-基]-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0134]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-噻唑-2-基-吡啶-3-甲酰胺;
[0135]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(2-甲基-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0136]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(2,4-二甲氧基嘧啶-5-基)-6-氧代-吡啶-3-甲酰胺;
[0137]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-噻唑-5-基-吡啶-3-甲酰胺;
[0138]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(2-甲氧基嘧啶-5-基)-6-氧代-吡啶-3-甲酰胺;
[0139]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(6-甲氧基-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0140]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(1-异丙基吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0141]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(1-环丙基吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0142]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-[(3r)-1-甲基吡咯烷-3-基]-6-氧代-吡啶-3-甲酰胺;
[0143]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-[(3s)-1-甲基吡咯烷-3-基]-6-氧代-吡啶-3-甲酰胺;
[0144]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(1,5-二甲基吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0145]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(3-环丙基-1-甲基-吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0146]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3-氰基苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0147]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-[1-(环丙基甲基)吡唑-4-基]-6-氧代-吡啶-3-甲酰胺;
[0148]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-[1-(2-甲氧基乙基)吡唑-4-基]-6-氧代-吡啶-3-甲酰胺;
[0149]
·
1-(1-叔丁基吡唑-4-基)-n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-吡啶-3-甲酰胺;
[0150]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0151]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1-甲基-1h-吡唑-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0152]
·
1-甲基-6-氧代-n-[4-(三氟甲氧基)苯基]吡啶-3-甲酰胺
[0153]
·
n-[4-(氯二氟甲氧基)苯基]-1-[1-(二氟甲基)-1h-吡唑-4-基]-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0154]
·
n-[4-(氯二氟甲氧基)苯基]-6-氧代-1-(喹喔啉-5-基)-1,6-二氢吡啶-3-甲酰胺;
[0155]
·
n-[4-(氯二氟甲氧基)苯基]-1-(4-甲氧基苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0156]
·
1-(5-氯-1-甲基-1h-吡唑-4-基)-n-[4-(氯二氟甲氧基)苯基]-6-氧代-1,6-二
氢吡啶-3-甲酰胺;
[0157]
·
n-[4-(氯二氟甲氧基)苯基]-1-(2,3-二氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0158]
·
n-[4-(氯二氟甲氧基)苯基]-1-(2,3-二甲氧基苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0159]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二氢-1h-2-苯并吡喃-6-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0160]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1,3-二氢-2-苯并呋喃-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0161]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3-甲氧基苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0162]
·
n-[4-(氯二氟甲氧基)苯基]-1-(2,3-二氢-1,4-苯并二英-6-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0163]
·
n-[4-(氯二氟甲氧基)苯基]-1-(2-氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0164]
·
n-[4-(氯二氟甲氧基)苯基]-6-氧代-1-(喹喔啉-6-基)-1,6-二氢吡啶-3-甲酰胺;
[0165]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二氢-1h-2-苯并吡喃-7-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0166]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二氢-1h-2-苯并吡喃-8-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0167]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二氢-1h-2-苯并吡喃-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0168]
·
n-[4-(氯二氟甲氧基)苯基]-1-(4-氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0169]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,4-二甲氧基苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0170]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1-甲基-1h-1,2,3-苯并三唑-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0171]
·
1-(3-氯-1-甲基-1h-吡唑-4-基)-n-[4-(氯二氟甲氧基)苯基]-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0172]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3,5-二氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0173]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3-氟苯基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0174]
·
1-(1,3-苯并唑-4-基)-n-[4-(氯二氟甲氧基)苯基]-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0175]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-[4-(2-氧代氮杂环丁烷-1-基)苯基]吡啶-3-甲酰胺;
[0176]
·
6-氧代-1-(嘧啶-5-基)-n-[4-(三氟甲氧基)苯基]-1.6-二氢吡啶-3-甲酰胺;
[0177]
·
n-[4-(氯二氟甲氧基)苯基]-6-氧代-1-(嘧啶-5-基)-1,6-二氢吡啶-3-甲酰胺;
[0178]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(1-甲基吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0179]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-(3-吡啶基)吡啶-3-甲酰胺;
[0180]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-氟-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0181]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-氰基-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0182]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-苯基-吡啶-3-甲酰胺;
[0183]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-甲基-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0184]
·
n-[4-[氯(二氟)甲氧基]苯基]-5-甲氧基-6-氧代-1-嘧啶-5-基-吡啶-3-甲酰胺;
[0185]
·
n-[4-[氯(二氟)甲氧基]苯基]-5-甲基-6-氧代-1-嘧啶-5-基-吡啶-3-甲酰胺;
[0186]
·
n-[4-(氯二氟甲氧基)苯基]-5-(吗啉-4-基)-6-氧代-1-(嘧啶-5-基)-1,6-二氢吡啶-3-甲酰胺;
[0187]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-甲氧基-3,吡啶基)-6-氧代-吡啶,3-甲酰胺;
[0188]
·
n-[4-[氯(二氟)甲氧基]苯基]-6-氧代-1-[5-(三氟甲基)-3-吡啶基]吡啶-3-甲酰胺;
[0189]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-氯-3-吡啶基)-6-氧代-吡啶-3-甲酰胺;
[0190]
·
n-[4-(氯二氟甲氧基)苯基]-1-{咪唑并[1,2-b]哒嗪-3-基}-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0191]
·
n-[4-(氯二氟甲氧基)苯基]-6
′‑
甲基-2-氧代-2h-[1,3
′‑
联吡啶]-5-甲酰胺;
[0192]
·
n-[4-(氯二氟甲氧基)苯基]-5
′‑
(二氟甲基)-2-氧代-2h-[1,3
′‑
联吡啶]-5-甲酰胺;
[0193]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1,5-萘啶-3-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0194]
·
n-[4-(氯二氟甲氧基)苯基]-1-(3-甲基-4-氧代-3,4-二氢喹唑啉-7-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0195]
·
n-[4-(氯二氟甲氧基)苯基]-1-{2h,3h-[1,4]二英[2,3-b]吡啶-7-基}-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0196]
·
n-[4-(氯二氟甲氧基)苯基]-1-{1-甲基-1h-吡唑并[4,3-b]吡啶-6-基}-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0197]
·
n-[4-(氯二氟甲氧基)苯基]-1-{2-甲基-2h-吡唑并[4,3-b]吡啶-6-基}-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0198]
·6′‑
氯-n-[4-(氯二氟甲氧基)苯基]-2-氧代-2h-[1,3
′‑
联吡啶]-5-甲酰胺;
[0199]
·
n-[4-(氯二氟甲氧基)苯基]-1-{2h,3h-呋喃并[2,3-b]吡啶-5-基}-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0200]
·2′‑
氯-n-[4-(氯二氟甲氧基)苯基]-2-氧代-2h-[1,3
′‑
联吡啶]-5-甲酰胺;
[0201]
·
n-[4-(氯二氟甲氧基)苯基]-2-氧代-6
′‑
(三氟甲基)-2h-[1.3
′‑
联吡啶]-5-甲
酰胺;
[0202]
·
n-[4-(氯二氟甲氧基)苯基]-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0203]
·
n-[4-(氯二氟甲氧基)苯基]-1-(完-4-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0204]
·
5-{[4-(氯二氟甲氧基)苯基]氨基甲酰基}-2-氧代-2h-[1.3
′‑
联吡啶]-1
′‑‑1′‑
醇盐;
[0205]
·
n-[4-(氯二氟甲氧基)苯基]-5-氰基-1-(1-甲基-1h-吡唑-4-基)-6-氧代-1.6-二氢吡啶-3-甲酰胺;
[0206]
·
n-[4-(氯二氟甲氧基)苯基]-6-氧代-5-(1h-吡唑-3-基)-1,6-二氢吡啶-3-甲酰胺;
[0207]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-5-(3-吡啶基)吡啶-3-甲酰胺;
[0208]
·
n-[4-(氯二氟甲氧基)苯基]-1-甲基-6-氧代-5-(嘧啶-5-基)-1,6-二氢吡啶-3-甲酰胺;
[0209]
·
n-[4-(氯二氟甲氧基)苯基]-1-甲基-5-(1-甲基-1h-吡唑-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0210]
·
n-[4-(氯二氟甲氧基)苯基]-5-(1-乙基-1h-吡唑-3-基)-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0211]
·
n-[4-(氯二氟甲氧基)苯基]-1-甲基-5-(1-甲基-1h-吡唑-3-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺;
[0212]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-5-(1-甲基吡唑-4-基)-6-氧代-吡啶-3-甲酰胺;
[0213]
·
n-[4-[氯(二氟)甲氧基]苯基]-5-(5-氟-3-吡啶基)-1-甲基-6-氧代-吡啶-3-甲酰胺;
[0214]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-5-苯基-吡啶-3-甲酰胺;
[0215]
·
n-[4-(氯二氟甲氧基)苯基]-1-(1-甲基-1h-吡唑-4-基)-2-氧代-1,2-二氢-[3,3
′‑
联吡啶]-5-甲酰胺;
[0216]
·
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-5-吡唑-1-基-吡啶-3-甲酰胺;
[0217]
·
n-[4-[氯(二氟)甲氧基]苯基]-5-咪唑-1-基-1-甲基-6-氧代-吡啶-3-甲酰胺;
[0218]
·
n-[4-(氯二氟甲氧基)苯基]-2-氧代-6-(1h-吡唑-5-基)-1,2-二氢吡啶-4-甲酰胺;
[0219]
·
2-氧代-6-(嘧啶-5-基)-n-[4-(三氟甲氧基)苯基]-1,2-二氢吡啶-4-甲酰胺;
[0220]
·
n-[4-(氯二氟甲氧基)苯基]-1-甲基-6-(1-甲基-1h-吡唑-4-基)-2-氧代-1,2-二氢吡啶-4-甲酰胺。
[0221]
本发明的化合物可包括同位素标记和/或同位素富集形式的化合物。本文中本发明的化合物可在构成这样的化合物的一个或更多个原子处包含非天然比例的原子同位素。可并入到所公开化合物中的同位素的一些实例包括氢、碳、氮、氧、磷、硫、氯的同位素,例如2h、3h、
11
c、
13
c、
14
c、
13
n、
15
o、
17
o、
32
p、
35
s、
18
f、
36
cl。
[0222]
本发明的化合物可原样使用,或在适当时作为其药理学上可接受的盐(酸或碱加
成盐)使用。下文提及的药理学上可接受的加成盐意指包含化合物能够形成的具有治疗活性的无毒酸和碱加成盐形式。通过用合适的酸处理碱形式,可将具有碱性特性的化合物转化为其药理学上可接受的酸加成盐。一些示例性酸包括无机酸,例如氯化氢、溴化氢、碘化氢、硫酸、磷酸;以及有机酸,例如甲酸、乙酸、丙酸、羟基乙酸、乳酸、丙酮酸、乙醇酸、马来酸、丙二酸、草酸、苯磺酸、甲苯磺酸、甲磺酸、三氟乙酸、富马酸、琥珀酸、苹果酸、酒石酸、柠檬酸、水杨酸、对氨基水杨酸、扑酸(pamoic acid)、苯甲酸、抗坏血酸等。一些示例性碱加成盐形式是钠盐、钾盐、钙盐,以及具有可药用胺的盐,例如如氨、烷基胺、苄星(benzathine)和氨基酸,例如如精氨酸和赖氨酸。本文中使用的术语加成盐还包含化合物及其盐能够形成的溶剂合物,例如水合物、醇化物(alcoholate)等。
[0223]
在整个本公开内容中,给定的化学式或命名还将涵盖其所有可药用盐、溶剂合物、水合物、n-氧化物和/或前药形式。应当理解,本发明的化合物包括化合物式的任何和所有水合物和/或溶剂合物。应理解,在化合物的多种物理形式中,某些官能团,例如羟基、氨基等基团与水和/或多种溶剂形成复合物和/或配位化合物。因此,上式应理解为包含并代表那些多种水合物和/或溶剂合物。
[0224]
本发明的化合物还包含互变异构形式。互变异构形式由单键与相邻双键的交换以及伴随的质子迁移而产生。互变异构形式包括质子移变互变异构体,其为具有相同经验式和总电荷的异构质子化状态。示例性质子移变互变异构体包括酮-烯醇对、酰胺-亚胺酸对、内酰胺-内酰亚胺对、酰胺-亚胺酸对、烯胺-亚胺对和其中质子可占据杂环体系的两个或更多个位置的环状形式,例如1h-和3h-咪唑、1h、2h-和4h-1,2,4-三唑、1h-和2h-异吲哚以及1h-和2h-吡唑。互变异构形式可通过适当的取代处于平衡中或空间锁定成一种形式。
[0225]
本文中所述的化合物可以是不对称的(例如,具有一个或更多个立构中心)。除非另外指明,否则意指所有立体异构体,例如对映体和非对映体。包含不对称的取代的碳原子的本发明的化合物可以以光学活性或外消旋形式分离。如何由光学活性起始物料制备光学活性形式的方法是本领域已知的,例如通过拆分外消旋混合物或通过立体选择性合成。烯烃、c=n双键等的许多几何异构体也可存在于本文中所述的化合物中,并且所有这样的稳定的异构体均在本发明中考虑。描述了本发明化合物的顺式-和反式-几何异构体,并且可将其作为异构体的混合物或作为经分离的异构体形式进行分离。
[0226]
在包含不对称碳原子的化合物的情况下,本发明涉及d型、l型和d,l混合物,并且在存在多于一个不对称碳原子的情况下,本发明还涉及非对映体形式。包含不对称碳原子且通常作为外消旋体产生的那些本发明化合物可以以已知方式例如使用光学活性酸分离成光学活性异构体。然而,也可从一开始就使用光学活性起始物料,随后获得相应的光学活性或非对映体化合物作为终产物。
[0227]
术语“前药”是指可在生理条件下或通过溶剂解转化为本发明的生物活性化合物的化合物。当向有此需要的对象施用时,前药可以是无活性的,但在体内转化为本发明的活性化合物。前药通常在体内快速转化以产生本发明的母体化合物,例如通过在血液中水解。前药化合物通常在哺乳动物生物体中提供溶解度、组织相容性或延迟释放的优点(参见silverman,r.b.,the organic chemistry of drug design and drug action,第2版,elsevier academic press(2004),第498至549页)。本发明化合物的前药可通过以将修饰在常规操作中或在体内裂解成本发明母体化合物这样的方式对本发明化合物中存在的官
能团,例如羟基、氨基或巯基进行修饰来制备。前药的一些实例包括但不限于羟基官能团的乙酸酯、甲酸酯和琥珀酸酯衍生物或者氨基官能团的氨基甲酸苯酯衍生物。
[0228]
本发明的另一个目的涉及用于治疗的本发明的化合物。
[0229]
本发明的化合物可用作c-abl的抑制剂。因此,其可用于治疗或预防其中c-abl的抑制是有益的医学病症(病症或疾病)。因此,提供了用于治疗或预防对c-ab1抑制有响应的疾病或病症的方法,其包括向对象施用治疗有效量的本发明的化合物。虽然本发明的化合物可适合于预防一系列疾病和病症,但优选其用于治疗所述疾病和病症。因此,优选该方法用于治疗疾病或病症,并因此该方法包括向有此需要的对象施用治疗有效量的本发明的化合物。
[0230]
本文中使用的术语“治疗”可包括预防指定的障碍或病症,或者一旦障碍已确立就减轻或消除该障碍。术语“预防”是指预防指定的障碍或病症。
[0231]
通过c-abl抑制可治疗或可预防的一系列疾病和病症是公知的。因此,本发明的化合物可用于治疗或预防该一系列疾病或病症。这包括神经退行性病症、癌症、朊病毒病、病毒感染、糖尿病、炎性疾病例如肺纤维化、急性胰腺炎(优选重症急性胰腺炎)、肺动脉高压或者骨骼或肌营养不良。优选地,疾病是神经退行性病症或癌症。
[0232]
可治疗或可预防的神经退行性病症包括但不限于阿尔茨海默病(alzheimer disease)、唐氏综合征(down’s syndrome)、额颞痴呆、进行性核上性麻痹、皮克病(pick’s disease)、尼曼-皮克病(niemann-pick disease)、帕金森病(parkinson’s disease)、亨廷顿病(huntington’s disease,hd)、齿状核红核苍白球丘脑下核萎缩(dentatorubropallidoluysian atrophy)、肯尼迪病(kennedy’s disease)、和脊髓小脑性共济失调、脆性x(雷特(rett’s))综合征、脆性xe智力低下、弗里德赖希共济失调(friedreich’s ataxia)、强直性肌营养不良、脊髓小脑性共济失调8型和脊髓小脑性共济失调12型、亚历山大病(alexander disease)、阿尔珀斯病(alper’s disease)、肌萎缩侧索硬化(als)、共济失调毛细血管扩张症、巴藤病(batten disease)、卡纳万病(canavan disease)、科凯恩综合征(cockayne syndrome)、皮质基底节变性、克罗伊茨费尔特-雅各布病(creutzfeldt-jakob disease)、缺血性卒中、克拉伯病(krabbe disease)、路易体痴呆(lewy body dementia)、多发性硬化、多系统萎缩、佩利措伊斯-梅茨巴赫病(pelizaeus-merzbacher disease)、皮克病、原发性侧索硬化、雷弗素姆氏病(refsum’s disease)、山德霍夫病(sandhoff disease)、希尔德病(schilder’s disease)、脊髓损伤、脊髓性肌萎缩症、斯蒂尔-理查森-奥尔谢夫斯基病(steele-richardson-olszewski disease)和脊髓痨。
[0233]
在可治疗或可预防的神经退行性病症中,最值得注意的是肌萎缩侧索硬化(als)和帕金森病。最优选地,神经退行性病症是als。
[0234]
可治疗或可预防的癌症包括但不限于白血病。
[0235]
在可治疗或可预防的癌症中,最值得注意的是慢性髓性白血病(cml)、急性淋巴母细胞白血病(all)、急性髓性白血病(aml)和混合表型急性白血病(mpal)、或其任何中枢神经系统(cns)转移。最优选地,癌症是cml或all。
[0236]
因此,本发明包括本发明化合物在制备用于治疗或预防疾病或病症(例如上述神经退行性病症和癌症)的药物中的用途。本发明还涉及用于治疗疾病或病症(例如上述神经退行性病症和癌症)的本发明的化合物。
[0237]
本发明的化合物可用作独立治疗或与其他c-abl抑制剂结合使用。可与本发明的化合物结合使用的c-abl抑制剂的实例是尼洛替尼、帕纳替尼、达沙替尼、博舒替尼及其混合物。
[0238]
本文中描述的方法包括其中对象被确定为需要特定的所述治疗的那些。确定需要这样的治疗的对象可以是在对象或健康护理专业人员的判断中,并且可以是主观的(例如意见)或客观的(例如可通过测试或诊断方法测量)。
[0239]
在另一些方面中,本文中的方法包括还包括监测对治疗施用有响应的对象的那些。这样的监测可包括对对象组织、流体、标本、细胞、蛋白质、化学标志物、遗传物质等的定期取样,作为治疗方案的标志物或指示物。在另一些方法中,通过评估用于这样的治疗的适合的相关标志物或指示物,将对象预筛选或确定为需要这样的治疗。
[0240]
本发明提供了监测治疗进展的方法。该方法包括在患有或易患有本文中描述的病症或其症状的对象中确定诊断标志物(标志物)(例如,本文中描述的由本文中的化合物调节的任何靶标或细胞类型)的水平或诊断测量(例如,筛选、测定)的步骤,其中已向对象施用了足以治疗疾病或其症状的治疗量的本文中的化合物。可将该方法中确定的标志物水平与健康正常对照中或其他患病患者中的标志物已知水平进行比较以确立对象的疾病状态。优选地,在晚于确定第一水平的时间点确定对象中标志物的第二水平,并比较这两个水平以监测疾病进程或治疗效力。在某些优选实施方案中,对象中标志物的治疗前水平在开始根据本发明的治疗之前确定;然后可将该标志物的治疗前水平与治疗开始之后对象中的标志物水平进行比较,以确定治疗的效力。
[0241]
对象中的标志物或标志物活性的水平可至少一次确定。标志物水平的比较,例如,与先前或随后从同一患者、另一患者或正常对象获得的标志物水平的另一测量的比较,可用于确定根据本发明的治疗是否具有期望的作用,并从而允许适当地调节剂量水平。标志物水平的确定可使用本领域已知的或本文中所述的任何合适的取样/表达测定方法进行。优选地,首先从对象中移出组织或流体样品。合适的样品的一些实例包括血液、尿、组织、口腔或颊细胞以及包含根部的毛发样品。另一些合适的样品将是本领域技术人员已知的。样品中蛋白质水平和/或mrna水平(例如标志物水平)的确定可使用本领域已知的任何合适的技术进行,包括但不限于酶免疫测定、is elisa、放射性标记/测定技术、印迹/化学发光方法、实时pcr等。
[0242]
对于临床应用,本文中公开的化合物被配制成用于多种施用模式的药物组合物(或制剂)。应当理解,本发明的化合物可与生理学上可接受的载体、赋形剂和/或稀释剂(即这些中的一种、两种或所有三种)一起施用。本文中公开的药物组合物可通过任何合适的途径施用,优选通过经口、经直肠、经鼻、表面(包括口含和舌下)、舌下、经皮、鞘内、经黏膜或肠胃外(包括皮下、肌内、静脉内和皮内)施用。其它制剂可方便地以单位剂型(例如,片剂和持续释放的胶囊剂)和以脂质体存在,并且可通过药学领域公知的任何方法来制备。药物制剂通常通过使活性物质或其可药用盐与常规的可药用载体、稀释剂或赋形剂混合来制备。赋形剂的一些实例是水、明胶、阿拉伯树胶、乳糖、微晶纤维素、淀粉、羟基乙酸淀粉钠、磷酸氢钙、硬脂酸镁、滑石、胶体二氧化硅等。这样的制剂还可包含其他药理学活性剂和常规添加剂,例如稳定剂、润湿剂、乳化剂、矫味剂、缓冲剂等。通常来说,活性化合物的量为按制剂的重量计0.1至95%,优选地为按用于肠胃外用制剂的重量计0.2至20%,并且更优选地为
c5炔基、c
3-c4炔基、c3炔基、c
4-c6炔基、c
4-c5炔基、c4炔基、c
5-c6炔基、c5炔基和c6炔基。“c
2-c6炔基”的一些实例包括2-丙炔基、2-丁炔基、3-丁炔基、2-戊炔基、3-甲基-4-戊炔基、2-己炔基、5-己炔基等。
[0253]
术语“c
1-c6烷氧基”表示-o-(c
1-c6烷基),其中c
1-c6烷基如上文所定义并且通过氧原子与化合物的其余部分连接。“c
1-c6烷氧基”的一些实例包括甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基以及直链和支链戊氧基和己氧基。
[0254]
术语“卤代”意指卤素原子,除非另有说明,否则优选f、cl、br和i,更优选f和cl,并且最优选f。
[0255]
术语“氧代”表示与氧原子(=o)的双键。这通常形成酮基或醛基,前者可以形成另外官能团(例如羧酸、酯或酰胺)的一部分。
[0256]
术语“6至10元芳基”表示包含6至10个环原子的稳定的芳族单环或稠合双环烃环体系。术语“6至10元芳基”包括稠合双环环体系,其中一个环是部分不饱和或完全饱和的,其中与分子的其余部分的连接点在芳族环上。“6至10元芳基”的实例包括苯基、茚基、萘基、萘、1,2,3,4-四氢萘基和茚满基。
[0257]
术语“5至10元杂芳基”表示具有5至10个环原子的稳定的芳族单环或稠合双环杂芳族环体系,其中1至9个环原子为碳并且一个或更多个环原子选自氮、硫和氧。“5至10元杂芳基”的一些实例包括呋喃基、吡咯基、噻吩基、唑基、异唑基、咪唑基、噻唑基、异噻唑基、吡啶基、嘧啶基、四唑基、喹唑啉基、吲哚基、二氢吲哚基、异吲哚基、异二氢吲哚基、吡唑基、哒嗪基、吡嗪基、喹啉基、喹喔啉基、噻二唑基、苯并呋喃基、2,3-二氢苯并呋喃基、1,3-苯并二氧杂环戊烯基、1,4-苯并二氧杂环己烯基(1,4-benzodioxinyl)、2,3-二氢-1,4-苯并二氧杂环己烯基、苯并噻唑基、苯并咪唑基、苯并噻二唑基、苯并三唑基、色满基和四氢喹啉。术语“5至10元芳基”包括稠合双环环体系,其中一个环是部分不饱和或完全饱和的,其中与分子的其余部分的连接点在芳族环上,例如在以下实例中:
[0258][0259]
术语“5元杂芳基”表示具有5个环原子的单环“5至10元杂芳基”,其中1至4个环原子为碳并且一个或更多个环原子选自氮、硫和氧。“5元杂芳基”的实例包括吡咯基和呋喃基。
[0260]
术语“6元杂芳基”表示具有6个环原子的单环“5至10元杂芳基”,其中1至5个环原子为碳并且一个或更多个环原子选自氮、硫和氧。“6元杂芳基”的实例包括吡啶基和嘧啶基。
[0261]
术语“4至10元杂环”表示具有5至10个环原子的非芳族单环或稠合双环的完全饱和或部分不饱和的环体系,其中1至9个环原子是碳并且一个或更多个环原子选自氮、硫和氧。当存在时,硫原子可以是氧化形式(即s=o的双基或o=s=o的双基)。术语“4至10元杂环”包括稠合双环环体系,其中一个环是芳族的,其中与分子的其余部分的连接点在非芳族环上。“4至10元杂环”的实例包括氮杂环丁烷基、哌啶基、四氢吡喃基、四氢呋喃基、氧杂环
丁烷基、氮杂基(azepinyl)、氮杂环丁烷基、吡咯烷基、吗啉基、咪唑啉基、咪唑烷基、硫代吗啉基、吡喃基、二烷基、哌嗪基、高哌嗪基和5,6-二氢-4h-1,3-嗪-2-基。
[0262]
术语“5元杂环”表示其中环体系具有5个环原子的单环“4至10元杂环”,其中1至4个环原子是碳并且一个或更多个环原子选自氮、硫和氧。“5元杂环”的实例包括四氢呋喃基和吡咯烷基。
[0263]
术语“6元杂环”表示其中环体系具有6个环原子的单环“4至10元杂环”,其中1至5个环原子是碳并且一个或更多个环原子选自氮、硫和氧。“6元杂环”的实例包括哌啶基和吗啉基。
[0264]“有效量”是指赋予所治疗对象治疗作用的本发明的化合物的量。治疗作用可以是客观的(即,通过一些测试或标志物可测量的)或主观的(即,对象给出对作用的指示或感觉到作用)。
[0265]
本文中使用的术语“施用”或其变化形式意指用于本文中公开的化合物的施用途径。一些示例性施用途径包括但不限于经口、静脉内、腹膜内、动脉内和肌内。优选的施用途径可取决于多种因素变化,例如包含本文中公开的化合物的药物组合物的组分、潜在或实际疾病的部位和疾病的重症程度。
[0266]
术语“对象”和“患者”在本文中可互换使用。它们是指可受疾病或病症折磨或易于患有疾病或病症但是可患有或可未患有疾病或病症的人或其他哺乳动物(例如,小鼠、大鼠、兔、狗、猫、牛、猪、绵羊、马或灵长类)。优选对象是人。
[0267]
本发明的化合物可通过名称或化学结构来公开。本文中的化合物使用openeye命名约定命名。如果化合物的名称与其相关化学结构之间存在差异,则以化学结构为准。
[0268]
现将通过以下非限制性实施例进一步说明本发明。以下具体实施例应被解释为仅是说明性的,并且不以任何方式限制本公开内容的其余部分。无需进一步阐述,认为本领域技术人员可基于本文中所述以其最大程度地利用本发明。本文中引用的所有参考文献和出版物通过引用整体并入于此。
[0269]
本发明化合物的制备
[0270]
本文中公开的式(i)化合物可以通过常规方法或以与常规方法类似的方法来制备。根据本发明的实例的中间体和化合物的制备可以特别地通过以下方案阐明。本文方案中结构变量的限定与本文描述的式中相应位置的变量的限定相称。
[0271]
方案1.用于制备式(ia)化合物的一般合成路线
[0272][0273]
其中r1、r2、r3、r4、r7和r8如式(i)中所限定并且m为na或li。
[0274]
其中a为方案1中描述的吡啶酮区域异构体的通式(i)化合物(通式(ia)指定的化合物)可以通过多种路线容易地制备。例如,通式(ia-i)的吡啶酮可以用r4i烷基碘进行n-烷基化,形成通式(ia-ii)化合物,其进而可以进行酯水解以得到通式(ia-iii)化合物,然后与wnh2苯胺进行酰胺偶联以得到通式(ia)化合物。或者,通式(ia-ii)化合物也可以通过用r4nh2胺使通式(ia-iv)香豆灵酸酯开环然后缩合-环化来制备。或者,通式(ia)化合物可以由式(ia-v)化合物通过包括烷基化、chan-lam或ullmann反应的标准化学方法来制备。式(ia-v)化合物可以由通式(ia-vi)5-溴-6-甲氧基烟酸酯使用标准交叉偶联反应例如suzuki和buchwald反应以得到通式(ia-vii)6-甲氧基烟酸酯,然后用酸处理以得到通式(ia-viii)化合物,随后与wnh2苯胺进行酰胺偶联来制备。如果需要,可以使用标准保护基策略来促进方案1中概述的合成。
[0275]
任选地,式(ia)化合物可以在一个或更多个合成步骤中转化成另一种式(ia)化合物。
[0276]
方案2.用于制备式(ib)化合物的一般合成路线
[0277][0278]
其中r1、r2、r5、r7和r8如式(i)中所限定。
[0279]
其中a为方案2中描述的吡啶酮区域异构体并且r6为h的通式(i)化合物(通式(ib)指定的化合物)可以通过多种路线容易地制备。例如,通式(ib-i)异烟酸可以与wnh2苯胺进行酰胺形成以得到通式(ib-ii)异烟酰胺,然后通过suzuki反应以得到通式(ib-iii)化合物。然后可以将通式(ib-iii)化合物用酸例如hcl处理以得到通式(ib)化合物。或者,通式(ib-iii)化合物可以由通式(ib-iv)异烟酸通过用wnh2苯胺进行酰胺形成以得到通式(ib-v)异烟酰胺,然后进行suzuki反应以得到通式(ib-vi)化合物,随后用甲醇钠处理来制备。如果需要的话,可以使用标准保护基策略来促进方案2中概述的合成。
[0280]
任选地,式(ib)化合物可以在一个或更多个合成步骤中转化成另一种式(ib)化合物。
[0281]
方案3.用于制备式(ic)化合物的一般合成路线
sons(1994);和l.paquette,ed.,encyclopedia of reagents for organic synthesis,john wiley and sons(1995)及其后续版本中描述的那些。
[0290]
现将通过以下非限制性实施例进一步说明本发明。以下具体实施例应被解释为仅是说明性的,并且不以任何方式限制本公开内容的其余部分。无需进一步阐述,认为本领域技术人员可基于本文中所述以其最大程度地利用本发明。本文中引用的所有参考文献和出版物通过引用整体并入于此。
[0291]
使用了以下缩写:
[0292]
[0293]
[0294]
[0295][0296]
实施例和中间体化合物
[0297]
除非另有说明,否则所有试剂均为商业级并且在未进一步纯化的情况下按原样使用。除非另有说明,否则使用试剂级溶剂。通过使用装有铝盖和隔膜的工艺瓶在biotage initiator系统上进行微波加热来促进反应。使用配备有redisep或graceresolv二氧化硅和c18 rp柱的combiflash companion或combiflash rf系统进行制备型低压色谱。在具有uv检测器的gilson系统、具有200nm至400nm uv可变波长检测器和purlon质谱仪的teledyne isco accqprep hp125系统、或者配备有dad和质量检测器的agilent 1260 infinity系统上进行制备型rp hplc。用以下柱中的至少一者配备rp hplc系统:ace-5aq,100mm
×
21.2mm,5μm柱;phenomenex synergi hydro-rp 80a axia,100mm
×
21.2mm,4μm柱;ace superc18,100mm
×
21.2mm,5μm柱;redisep c18prep,250mm
×
50.0mm,5μm柱;或者waters sunfire c18 obd prep column,5μm,19mm
×
100mm以及sunfire c18 prep guard cartridge,10μm,19mm
×
10mm。收集最纯的级分、将其浓缩并且在真空下干燥。在纯度分析之前,通常将化合物在40℃与60℃之间的真空烘箱中干燥。当用上标$标记时,标出的产量包括纯度系数。除非另有说明,否则在室温下进行反应。使用openeye规则自动命名化合物。
[0298]
通过lcms、hplc和uplc进行化合物分析。使用以下收集lcms数据:连接有waters zq质谱仪的agilent 1100 hplc系统、连接有acquity qda质量检测器的waters acquity h级uplc、具有dad\elsd和agilent lc\msd vl(g1956a)、sl(g1956b)质谱仪的agilent 1100系列lc/msd系统;或者具有dad\elsd和agilent lc\msd sl(g6130a)、sl(g6140a)质谱仪的agilent 1200系列lc/msd系统。报告的质量值对应于具有氢添加的[mh]
+
或钠添加的[mna]
+
的母体分子。在具有dad的agilent 1100系统、具有dad的agilent 1200系统或者具有dad的agilent 1290 infinity系统上收集hplc和uplc数据。分析型hplc或uplc系统利用以下柱
和方法:phenomenex kinetex xb-c18柱(1.7μm,2.1mm
×
100mm),在40℃下和以0.5ml/分钟,梯度为在水(
+
0.1%tfa)中的5%mecn(
+
0.085%tfa)持续1分钟,在8.0分钟内5%至100%,保持0.2分钟,然后再平衡0.8分钟,其中收集波长为200nm至300nm;phenomenex kinetex xb-c18柱(1.7μm,2.1mm
×
50mm),在40℃下和以0.8ml/分钟,梯度为在水(
+
0.1%tfa)中的5%mecn(
+
0.085%tfa)持续1分钟,在3.0分钟内5%至100%,保持0.2分钟,然后再平衡0.8分钟,其中收集波长为200nm至300nm;在phenomenex kinetex xb-c18柱(1.7μm,2.1mm
×
50mm),在40℃下和以0.8ml/分钟,梯度为在水(
+
0.1%tfa)中5%mecn(
+
0.085%tfa)持续1分钟,在3.0分钟内5%至100%,保持0.2分钟,然后再平衡0.8分钟,其中收集波长为200nm至300nm;zorbax sb-c18柱(1.8μm,4.6mm
×
15mm快速离析柱),在3.0ml/分钟下,梯度为在水(0.1%甲酸)中0%mecn(0.1%甲酸)持续0.01分钟,在1.5分钟内0%至100%,保持0.3分钟,然后再平衡0.2分钟;或者phenomenex kinetex xb-c18柱(1.7μm,2.1mm
×
50mm),在40℃下和0.8ml/分钟,梯度为在0.9分钟内在水(
+
0.1%tfa)中0%至20%mecn(
+
0.085%tfa),然后在0.1分钟内斜升至100%,保持0.2分钟,然后再平衡0.8分钟,其中收集波长为200nm至300nm。
[0299]
中间体1
[0300]
1-(2-吗啉代乙基)-6-氧代-吡啶-3-羧酸甲酯
[0301][0302]
使用微波反应器在110℃下将在mecn(4.0ml)中的6-氧代-1,6-二氢-3-吡啶羧酸甲酯(123mg,0.80mmol)、4-(2-氯乙基)吗啉、hcl(223mg,1.20mmol)、tabi(29.6mg,0.08mmol)和k2co3(332mg,2.40mmol)加热60分钟,过滤,然后在真空中浓缩。通过rp柱色谱进行纯化,得到作为无色胶状物的标题化合物(162mg,76.0%)。lcms(es
+
):267.1[mh]
+
。hplc:rt 0.64分钟,100%纯度。
[0303]
中间体2
[0304]
1-(1-甲基吡唑-3-基)-6-氧代-吡啶-3-羧酸甲酯
[0305][0306]
将在meoh(20ml)中的1-甲基-1h-吡唑-3-胺(2.19g,22.5mmol)和香豆灵酸甲酯(methyl coumalate)(2.16g,20.5mmol)搅拌20分钟,然后通过过滤收集沉淀物并将其在真空下,45℃下干燥4小时。将粗固体溶解在15%na2co3水溶液(40ml)中,加热至40℃至45℃,搅拌15分钟,冷却至室温,并且通过过滤收集固体材料,然后由ipa将其重结晶以得到作为灰白色固体的标题化合物(2.62g,57.3%)。lcms(es
+
):234.1[mh]
+
。
[0307]
中间体3至25
[0308]
类似于中间体2,通过用合适的伯胺使香豆灵酸甲酯开环,然后进行缩合环化来制备中间体3至25;参见下表1。
[0309]
表1:香豆灵酸甲酯与合适的伯胺的反应
[0310][0311]
[0312]
[0313]
[0314]
[0315]
[0316][0317]
中间体26
[0318]
1-(3,4-二甲氧基苯基)-6-氧代-吡啶-3-羧酸
[0319][0320]
将在无水meoh(10ml)中的香豆灵酸甲酯(503mg,3.25mmol)和3,4-二甲氧基苯胺(510mg,3.24mmol)回流2小时,冷却至室温,然后添加naoh(264mg,6.50mmol)并且将rm搅拌另外的12小时。将rm用水(40ml)进行处理,用etoac(20ml)和chcl3(20ml)进行洗涤。将水层使用10%hcl酸化至ph为2并且通过etoac(2
×
20ml)进行萃取。将合并的有机层经na2so4干燥并将溶剂蒸发。将粗产物通过rp hplc进行纯化以得到作为白色固体的标题化合物
(54.0mg,6.2%)。lcms(es
+
):276.0[mh]
+
。
[0321]
中间体27至47
[0322]
类似于中间体26,通过用合适的伯胺使香豆灵酸甲酯开环,然后进行缩合环化和随后的酯水解来制备中间体27至47;参见下表2。
[0323]
表2:用合适的伯胺使香豆灵酸甲酯开环,然后进行酯水解的反应
[0324][0325]
[0326]
[0327]
[0328][0329]
[0330]
中间体48
[0331]
5-溴-1-(1-甲基吡唑-4-基)-6-氧代-吡啶-3-羧酸
[0332][0333]
类似于中间体26,使用3-溴-2-氧代-2h-吡喃-5-羧酸甲酯代替香豆灵酸甲酯并且使用1-甲基-1h-吡唑-4-胺代替3,4-二甲氧基苯胺以得到作为黄色固体的标题化合物(9.54g,78.3%)来制备中间体48。lcms(es
+
):298.0[mh]
+
。
[0334]
中间体49
[0335]
1-(1,3-苯并坐-4-基)-6-氧代-吡啶-3-羧酸锂
[0336][0337]
将在1∶1 etoh∶水(10ml)中的中间体25(312mg,1.12mmol)和lioh(442mg,1.12mmol)在室温下进行搅拌直至tlc分析(etoac/10%meoh)表明了起始材料的完全消耗。将溶剂在减压下除去并且将剩余物在真空下干燥,从而得到作为黄色固体的标题化合物(244mg,78.4%),所述标题化合物在未纯化或表征的情况下在接下来的步骤中使用。
[0338]
中间体50
[0339]
6-甲氧基-5-吗啉代-吡啶-3-羧酸甲酯
[0340][0341]
将在无水二氧六环中的5-溴-6-甲氧基烟酸甲酯(3.04g,12.2mmol)和吗啉(2.13g,24.4mmol)用cs2co3(11.9g,36.6mmol)、pd2(dba)3(331mg,366μmol)和xantphos(421mg,732μmol)进行处理,然后加热至100℃过夜。将rm与水(50ml)进行混合,用etoac(3
×
50ml)进行萃取,将合并的有机萃取物在真空下浓缩以得到标题化合物,所述标题化合物在未进一步纯化或表征的情况下在接下来的步骤中使用。
[0342]
中间体51
[0343]
5-吗啉代-6-氧代-1h-吡啶-3-羧酸
[0344][0345]
将中间体50(1.03g,3.97mmol)和浓hcl水溶液(5.0ml)在回流下加热12小时,然后在真空下浓缩以得到标题化合物,所述标题化合物在未进一步纯化或表征的情况下在接下
来的步骤中使用。
[0346]
中间体52
[0347]
6-氧代-5-(1h-吡唑-5-基)-1h-吡啶-3-羧酸
[0348][0349]
将5-溴-6-甲氧基烟酸甲酯(3.04g,12.2mmol)、3-(四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1h-吡唑(3.08g,15.9mmol)、k2co3(6.73g,48.8mmol)和pd(dppf)cl2·
dcm(500mg,0.61mmol)在二氧六环-水(100ml,1∶1,v/v)中加热至90℃持续12小时。将rm用水(50ml)进行稀释,用etoac(2
×
30ml)进行萃取,并且将合并的有机层在真空中浓缩。将剩余物用浓hcl溶液(10ml)和thf(10ml)进行处理,在35℃至40℃下搅拌1小时,然后将预沉淀的固体过滤并干燥以得到作为灰色粉末的标题产物(922mg,36.7%)。lcms(es
+
):206.0[mh]
+
。
[0350]
一般的酰胺化步骤a、b、c、d和e
[0351][0352]
通过合适的苯胺与合适的羧酸的反应而在整个本发明中使用数个酰胺化步骤以制备多种中间体和示例的化合物。r1和r2如式(i)中限定并且z为如式(i)中限定的a或者z为制备a中使用的中间体。以下概述了一般性的、代表性的酰胺化方案a至e。
[0353]
一般的酰胺化步骤a:
[0354]
将合适的羧酸(25.8mmol)在0℃下混悬在无水dcm(100ml)和干燥dmf(200μl)中,然后逐滴添加cocl2(2.22ml,25.8mmol),并且将混合物在室温下搅拌2小时。添加dipea(9.00ml,51.7mmol)和合适的苯胺(25.8mmol)并将反应物在室温下搅拌18小时。小心地添加饱和nahco3水溶液(50ml)并且将混合物用dcm或etoac(3
×
50ml)进行萃取。将合并的有机萃取物在真空中浓缩并且典型地通过柱色谱、rp hplc、粉碎或结晶进行纯化。
[0355]
一般的酰胺化步骤b:
[0356]
将在二氧六环(20ml)中的合适的羧酸(5.91mmol)、合适的苯胺(8.87mmol)、edcl
·
hcl(2.27g,11.8mmol)、dmap(72.2mg,591μmol)和dipea(3.09ml,17.7mmol)在回流下进行搅拌直至通过lcmc显示反应完成,然后将其在真空中浓缩并且典型地通过柱色谱、rp hplc、粉碎或结晶进行纯化。
[0357]
一般的酰胺化步骤c:
[0358]
将合适的羧酸混悬在二氧六环(12ml)中,然后添加hatu(466mg,1.22mmol)和dipea(213μl,1.22mmol),并且将rm在80℃下搅拌15分钟。将合适的苯胺(284mg,1.47mmol)添加在二氧六环(1ml)中并且将rm在80℃下搅拌过夜,然后在真空中浓缩并且典型地通过柱色谱、rp hplc、粉碎或结晶进行纯化。
[0359]
一般的酰胺化步骤d:
[0360]
将在dmf(10ml)中的合适的羧酸(14.3mmol,1当量)和cdi(17.2mmol,1.2当量)在80℃下搅拌2小时,然后添加需要的苯胺(15.7mmol,1.1当量)并且将rm在80℃下搅拌过夜。
将rm冷却至室温,将dmf在真空下除去,将剩余物与水进行混合并且用etoac(3
×
20ml)进行萃取。将合并的有机萃取物经na2so4干燥、过滤并且在真空下浓缩,然后典型地通过柱色谱、rp hplc、粉碎或结晶进行纯化。
[0361]
一般的酰胺化步骤e:
[0362]
将pybop(253mg,1.12mmol,1.0当量)添加至在无水dmf(10ml)中的合适的羧酸(1.12mmol,1.0当量)、合适的苯胺(1.12mmol,1.0当量)和dipea(213mg,1.65mmol,1.5当量)的溶液。将rm在室温下搅拌过夜,然后用水(10ml)进行处理并且用etoac(2
×
10ml)进行萃取。将合并的有机层在na2so4下干燥,在真空下浓缩并且典型地通过柱色谱、rp hplc、粉碎或结晶进行纯化。
[0363]
中间体53至63
[0364]
通过与在合适的羧酸与合适的苯胺之间的一般的酰胺化步骤a至e类似的步骤制备中间体53至63;参见下表3。如果没有指定中间体,则合适的反应物是商业来源的。
[0365]
表3:酰胺化反应
[0366]
[0367]
[0368][0369]
中间体64
[0370]
5-溴-n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-吡啶-3-甲酰胺
[0371][0372]
将中间体55(1.70g,89.6%纯度,3.87mmol)、碘甲烷(241μl,3.87mmol)和k2co3(535mg,3.87mmol)在dmso(25ml)中搅拌18小时。将水(30ml)添加至快速搅拌的反应混合物,然后将反应混合物冷却至0℃至5℃并且搅拌10分钟。通过过滤收集固体材料,将其用水(3
×
20ml)进行洗涤,然后在50℃下的真空烘箱中干燥4小时以得到作为灰白色固体的标题化合物(1.58g,94.4%
$
)。lcms(es
+
):407.1[mh]
+
。hplc:rt 5.71分钟,94.2%纯度。
[0373]
中间体65
[0374]
2-氯-6-嘧啶-5-基-n-[4-(三氟甲氧基)苯基]吡啶-4-甲酰胺
[0375][0376]
在氩气下将中间体62(4.64g,13.0mmol)、(嘧啶-5-基)硼酸(813mg,6.53mmol)、25%k2co3水溶液(4.10g,29.5mmol)和pd(dppf)cl2·
dcm(114mg,0.13mmol)在二氧六环(20ml)中在90℃下搅拌4小时。将rm冷却至室温,与水(100ml)和etoac(100ml)进行混合,并且将分离的有机层经na2so4干燥并且在真空下浓缩。通过柱色谱(己烷/tbme)进行纯化,得到作为黄色固体的标题化合物(1.10g,35.4%)。lcms(es
+
):394.0[mh]
+
。
[0377]
中间体66
[0378]
2-甲氧基-6-嘧啶-5-基-n-[4-(三氟甲氧基)苯基]吡啶-4-甲酰胺
[0379][0380]
将meoh(0.12ml,3.00mmol)溶解在无水dmf(5.0ml)中,然后添加nah(在矿物油中的60%,61.0mg,1.50mmol),然后将所得混合物在室温下搅拌30分钟并且在60℃下搅拌30分钟。添加中间体65(203mg,0.51mmol)并且将rm在60℃搅拌下放置过夜。将rm用数滴acoh进行中和,在真空下浓缩,然后通过hplc进行纯化以得到作为白色固体的标题化合物(113mg,55.4%)。lcms(es
+
):390.0[mh]
+
。
[0381]
中间体67
[0382]
2-氯-n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-吡啶-4-甲酰胺
[0383]
[0384]
将中间体63(1.52g,4.30mmol)溶解在dmf(4.0ml)中并添加nah(在矿物油中的60%,213mg,4.70mmol)。将rm在室温下搅拌15分钟,然后添加碘甲烷(904mg,6.45mmol)。将rm在70℃下加热过夜,用水(10ml)进行稀释,然后通过过滤收集沉淀物。重结晶(1∶2 etoac∶己烷(hex)),得到作为浅棕色固体的标题化合物(484mg,31.5%)。lcms(es
+
):364.0[mh]
+
。
[0385]
中间体68
[0386]
n-[4-[氯(二氟)甲氧基]苯基]-3-碘-苯甲酰胺
[0387][0388]
类似于一般的酰胺化步骤c,使用3-碘苯甲酸作为酸反应物以得到作为浅棕色固体的标题化合物(3.56g,98.7%)来合成中间体68。lcms(es
+
):423.9[mh]
+
。uplc:rt 3.25分钟,98.1%纯度。
[0389]
中间体69
[0390]
3-碘-4-甲基-n-[4-(三氟甲氧基)苯基]苯甲酰胺
[0391][0392]
类似于一般的酰胺化步骤c,使用3-碘-4-甲基-苯甲酸作为酸反应物以得到作为白色固体的标题化合物(3.21g,74.7%
$
)来合成中间体69。lcms(es
+
):421.9[mh]
+
。uplc:rt 3.29分钟,92.7%纯度。
[0393]
中间体70
[0394]
5-(3-吡啶基)吡啶-3-羧酸甲酯
[0395][0396]
将5-溴吡啶-3-羧酸甲酯(2.08g,9.61mmol)在1,4-二氧六环(20ml)和水(1.5ml)中用n2脱气5分钟。添加3-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶(2.07g,10.1mmol)、pd(pph3)4(1.11g,0.96mmol)和k2co3(1.99g,14.4mmol),并将其在80℃下加热20小时。使混合物冷却至室温,将其过滤并在真空中浓缩。将粗材料用2m hcl(40ml)和etoac(160ml)进行处理,将水层分离并添加至etoac(200ml)中。将混合物用na2co3中和至ph为约7,并将相进行分离。将有机相干燥(mgso4)并在真空中浓缩以得到作为灰白色固体的标题化合物(1.78g,86.4%
$
)。lcms(es
+
):215.1[mh]
+
。uplc:rt 1.99分钟,98.3%纯度。
[0397]
中间体71
[0398]
5-(3-吡啶基)吡啶-3-羧酸
[0399][0400]
将中间体70(1.78g,8.30mmol)溶解在thf(10ml)和水(10ml)中,冷却至0℃,添加lioh(522mg,12.5mmol)并搅拌30分钟。将有机物蒸发,将水相用水(20ml)进行稀释并且用etoac(20ml)进行洗涤。将水相用khso4水溶液(1m)进行酸化直至ph为约4,通过过滤收集沉淀物并且将其用水进行洗涤以得到作为白色固体的标题化合物(929mg,55.9%
$
)。lcms(es
+
):201.1[mh]
+
。uplc:rt 1.20分钟,100%纯度。
[0401]
实施例1
[0402]
n-[4-氯(二氟)甲氧基]苯基]-1-(3-乙氧基-1-甲基-吡唑-4-基)-6-氧代-吡啶-3-甲酰胺
[0403][0404]
将中间体3(144mg,99.4%纯度,516μmol)和1m naoh水溶液(568μl,568μmol)在thf∶水(1∶1,4.0ml)中搅拌1小时。添加1m hcl水溶液(568μl,568μmol),然后将混合物在真空中浓缩并且在60℃下的真空烘箱中干燥过夜。类似于一般的酰胺化步骤c,将剩余物与4-[氯(二氟)甲氧基]苯胺(120mg,620μmol)进行反应。通过pr hplc进行纯化以及进行粉碎(meoh∶水),得到作为白色固体的标题化合物(62.1mg,27.3%
$
)。lcms(es
+
):439.0[mh]
+
。uplc:rt 5.80分钟,99.4%纯度。
[0405]
实施例2至24
[0406]
类似于实施例1,将中间体1、2和4至24进行酯水解,然后通过类似于一般的酰胺化步骤a至e将酰胺与合适的苯胺进行偶联来制备实施例2至24;参见下表4。
[0407]
表4:酯水解,然后酰胺偶联反应
[0408]
[0409]
[0410]
[0411]
[0412]
[0413]
[0414]
[0415]
[0416][0417]
实施例25
[0418]
n-[4-(氯二氟甲氧基)苯基]-1-(1-甲基-1h-吡唑-5-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺
[0419][0420]
类似于一般的酰胺化步骤d,将中间体27(248mg,1.14mmol)与4-[氯(二氟)甲氧基]苯胺(263mg,1,14mmol)进行反应。通过pr hplc进行纯化,得到作为白色固体的标题化合物(132mg,29.2%)。lcms(es
+
):395.2[mh]
+
。uplc:rt 5.38分钟,99.9%纯度。
[0421]
实施例26至49
[0422]
类似于实施例25,通过使用类似于一般的酰胺化步骤a至e的步骤将合适的羧酸与合适的苯胺进行酰胺偶联来制备实施例26至49;参见下表5。如果没有指定中间体,则使用商业反应物。
[0423]
表5:酰胺化反应
[0424][0425]
[0426]
[0427]
[0428]
[0429]
[0430]
[0431]
[0432]
[0433][0434]
实施例50
[0435]
6-氧代-1-(嘧啶-5-基)-n-[4-(三氟甲氧基)苯基]-1.6-二氢吡啶-3-甲酰胺
[0436][0437]
向meoh(10ml)中的(嘧啶-5-基)硼酸(398mg,3.22mmol)中顺序地添加cu(otf)2(1.17g,3.22mmol)和中间体54,然后添加吡啶(551μl,5.15mmol)。在配备有泡沫计数器的密封容器中将rm在25℃下搅拌16小时,然后将沉淀物滤出并将滤液在真空下浓缩。将剩余物溶解在etoac(20ml)中,用氨水溶液(2
×
50ml)进行洗涤,并且将有机层在真空下浓缩。通过hplc进行纯化,得到作为棕色固体的标题化合物(253mg,20.9%)。lcms(es
+
):377.0[mh]
+
。hplc:rt 4.95分钟,99.7%纯度。
[0438]
实施例51至60
[0439]
类似于实施例50,通过中间体53、56、57和60与合适的芳基或杂芳基硼酸酯或硼酸的chan-lam偶联来制备实施例51至60;参见下表6。
[0440]
表6:chan-lam反应
[0441]
[0442]
[0443]
[0444]
[0445][0446]
实施例61
[0447]
n-[4-[氯(二氟)甲氧基]苯基]-1-(5-甲氧基-3-吡啶基)-6-氧代-吡啶-3-甲酰胺
[0448][0449]
将在1,4-二氧六环(3.0ml)中的中间体53(200mg,97.8%纯度,622μmol)、cs2co2(621mg,1.91mmol)、dmeda(137μl,1.27mmol)和3-溴-5-甲氧基吡啶(239mg,1.27mmol)用n2脱气5分钟。添加cui(60.5mg,318μmol),将瓶密封并且在120℃下加热15小时。将rm添加至水(30ml)并用dcm(30ml)进行萃取。将有机相干燥(mgso4)并且在真空中浓缩。将剩余物通过rp柱色谱进行纯化以得到作为灰白色固体的标题化合物(53.5mg,20.4%
$
)。lcms(es
+
):422.0[mh]
+
。hplc:rt 5.40分钟,99.8%纯度。
[0450]
实施例62至75
[0451]
类似于实施例61,通过中间体53与合适的芳基或杂芳基卤化物的ullmann反应来制备实施例62至75;参见下表7。
[0452]
表7:ullmann反应
[0453][0454]
[0455]
[0456]
[0457]
[0458][0459]
实施例76
[0460]
n-[4-(氯二氟甲氧基)苯基]-1-甲基-6-氧代-1,6-二氢吡啶-3-甲酰胺
[0461][0462]
按照类似于实施例61的合成中使用的步骤的步骤,将实施例76作为来自中间体53与3-溴-5-氟-4-甲氧基吡啶之间的尝试的uiiman偶联的副产物分离。将实施例76分离,为白色固体(16.0mg,16.3%)。lcms(es
+
):329.1[mh]
+
。hplc:rt 2.55分钟,100%纯度。
[0463]
预期可以按照一般的酰胺化步骤c,类似于实施例26,使用4-[氯(二氟)甲氧基]苯胺代替4-(三氟甲氧基)苯胺来制造实施例76。
[0464]
实施例77
[0465]
n-[4-(氯二氟甲氧基)苯基]-1-(烷-4-基)-6-氧代-1,6-二氢吡啶-3-甲酰胺
[0466][0467]
将在dmf(5.0ml)中的中间体53(223mg,0.63mmol)、na2co3(134mg,1.26mmol)和4-碘四氢-2h-吡喃(164mg,0.76mmol)在室温下搅拌过夜,然后在etoac(30ml)与水(10ml)之间进行配分,将etoac层经na2so4干燥,过滤并蒸发至干燥,将剩余物通过制备型hplc进行纯化以得到作为白色固体的标题化合物(12.0mg,5.3%)。lcms(es
+
):399.0[mh]
+
。hplc:rt 6.24分钟,99.4%纯度。
[0468]
实施例78
[0469]
5-{[4-(氯二氟甲氧基)苯基]氨基甲酰基}-2-氧代-2h-[1.3
′‑
联吡啶]-1
′‑‑1′‑
醇盐
[0470][0471]
将实施例53(213mg,0.54mmol)溶解在dcm(5.0ml)中并添加mcpba(284mg,1.60mmol)。将所得混合物在室温下搅拌24小时,用1m naoh水溶液(2
×
10ml)进行洗涤,然后将有机层经na2so4干燥,在减压下浓缩并且通过制备型hplc进行纯化以提供作为黄色固体的标题化合物(27.0mg,12.3%)。lcms(es
+
):408.0[mh]
+
。hplc:rt 2.45分钟,95.8%纯度。
[0472]
实施例79
[0473]
n-[4-(氯二氟甲氧基)苯基]-5-氰基-1-(1-甲基-1h-吡唑-4-基)-6-氧代-1.6-二氢吡啶-3-甲酰胺
[0474][0475]
将cucn(94.0mg,1.05mmol)添加至中间体61(335mg,0.70mmol),然后将混合物在回流下加热10小时,冷却至80℃并倒入在水(10ml)中的nacn(214mg,4.37mmol)的溶液。在室温下搅拌1小时之后,将混合物用etoac(2
×
25ml)进行萃取,并且将有机相用盐水(10ml)进行洗涤,经na2so4干燥,在减压下浓缩并且通过rp hplc进行纯化以得到作为米色固体的标题化合物(104mg,34.1%)。lcms(es
+
):420.0[mh]
+
。uplc:rt 2.75分钟,99.6%纯度。
[0476]
实施例80
[0477]
n-[4-(氯二氟甲氧基)苯基]-6-氧代-5-(1h-吡唑-3-基)-1,6-二氢吡啶-3-甲酰
胺
[0478][0479]
类似于实施例25,使用中间体52代替中间体27,并且按照一般的酰胺化步骤d以得到作为白色固体的标题化合物(63.0mg,6.6%)来制备实施例80。lcms(es
+
):381.0[mh]
+
。hplc:rt 2.51分钟,99.2%纯度。
[0480]
实施例81
[0481]
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-5-(3-吡啶基)吡啶-3-甲酰胺
[0482][0483]
在n2下,110℃下将二氧六环(1.0ml)中的中间体64(100mg,94.2%纯度,231μmol)、吡啶-3-基硼酸(56.8mg,462μmol)、k2co3(95.8mg,693μmol)、pd(oac)2(10.4mg,46.2μmol)和pph3(30.3mg,116μmol)在密封管中加热18小时,然后将rm通过柱色谱进行纯化以得到作为米色固体的标题化合物(48.3mg,50.8%
$
)。lcms(es
+
):406.0[mh]
+
。uplc:rt 4.54分钟,98.7%纯度。
[0484]
实施例82至89
[0485]
类似于实施例81,通过中间体61和64与合适的芳基或杂芳基硼酸或硼酸酯的suzuki反应制备实施例82至89;参见下表8。
[0486]
表8:suzuki反应
[0487]
[0488]
[0489]
[0490][0491]
实施例90
[0492]
n-[4-[氯(二氟)甲氧基]苯基]-1-甲基-6-氧代-5-吡唑-1-基-吡啶-3-甲酰胺
[0493][0494]
向甲苯(4.0ml)中的中间体64(100mg,94.2%纯度,231μmol)、cui(8.80mg,46.2μ
mol)和k2co3(95.8mg,693μmol)的混合物添加反式n,n
′‑
二甲基环己烷-1,2-二胺(7.29μl,46.2μmol)和吡唑(31.5mg,462μmol),然后将rm在氮气下,110℃下加热66小时。将rm用dcm(5.0ml)稀释,过滤,然后通过柱色谱和rp hplc进行纯化以得到作为白色固体的标题化合物(21.6mg,23.5%
$
)。lcms(es
+
):394.8[mh]
+
。uplc:rt 5.81分钟,99.4%纯度。
[0495]
实施例91
[0496]
n-[4-[氯(二氟)甲氧基]苯基]-5-咪唑-1-基-1-甲基-6-氧代-吡啶-3-甲酰胺
[0497][0498]
类似于来自中间体64的实施例90,使用咪唑代替吡唑以得到作为米色固体的标题化合物(8.25mg,5.9%
$
)来制备实施例91。lcms(es
+
):395.0[mh]
+
。uplc:rt 4.53分钟,97.7%纯度。
[0499]
实施例92
[0500]
n-[4-(氯二氟甲氧基)苯基]-2-氧代-6-(1h-吡唑-5-基)-1,2-二氢吡啶-4-甲酰胺
[0501][0502]
将二氧六环-水(10ml,1∶1,v/v)中的中间体59(201mg,554μmol)、3-(四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1h-吡唑(140mg,720μmol)、k2co3(305mg,2.22mmol)和pd(dppf)cl2·
dcm(23.0mg,28.0μmol)加热至90℃持续12小时。将rm与水(5.0ml)进行混合,用etoac(2
×
10ml)进行萃取,并在真空中浓缩。将剩余物用浓hcl(5.0ml)和thf(5.0ml)进行处理并且在35℃至40℃下搅拌1小时。在冷却至室温之后,将rm用etoac(2
×
10ml)进行萃取,并且将合并的有机萃取物经mgso4干燥,在真空中浓缩并且通过rp hplc进行纯化以得到作为白色固体的标题化合物(12.0mg,5.7%)。lcms(es
+
):381.2[mh]
+
。uplc:rt 2.56分钟,99.2%纯度。
[0503]
实施例93
[0504]
2-氧代-6-(嘧啶-5-基)-n-[4-(三氟甲氧基)苯基]-1,2-二氢吡啶-4-甲酰胺
[0505][0506]
将中间体66(313mg,0.77mmol)溶解在acoh(2.0ml)中,添加浓hcl(2.0ml),并且将rm在50℃下搅拌12小时,然后在真空中浓缩并且通过rp hplc进行纯化以得到作为灰白色固体的标题化合物(14.0mg,3.1%)。lcms(es
+
):377.2[mh]
+
。hplc:rt 1.26分钟,100%纯
度。
[0507]
实施例94
[0508]
n-[4-(氯二氟甲氧基)苯基]-1-甲基-6-(1-甲基-1h-吡唑-4-基)-2-氧代-1,2-二氢吡啶-4-甲酰胺
[0509][0510]
将中间体67(152mg,0.41mmol)、1-甲基-4-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)-1h-吡唑(134mg,0.62mmol)、30%k2co3水溶液(284mg,2.07mmol)和pd(dppf)cl2·
dcm(9.1mg,12.4μmol)的混合物进行抽空并用氩气回填三次。添加二氧六环(2.0ml)并且将rm在100℃下搅拌过夜,然后冷却至室温,将有机层分离,在na2so4下进行干燥并蒸发。将粗产物通过rp hplc(水/mecn)进行纯化以得到作为白色固体的标题化合物(25.0mg,15.2%)。lcms(es
+
):409.2[mh]
+
,rt 5.40分钟,99.4%纯度。
[0511]
参照化合物
[0512]
参照例1
[0513]
n-[4-[氯(二氟)甲氧基]苯基]-6-[(3r)-3-羟基吡咯烷-1-基]-5-(1h-吡唑-5-基)吡啶-3-甲酰胺
[0514][0515]
从medchemexpress商购asciminib(cas:1492952-76-7)并且将其作为原样使用。
[0516]
参照例2
[0517]
n-[4-[氯(二氟)甲氧基]苯基]-3-嘧啶-5-基苯甲酰胺
[0518][0519]
通过微波将1,4-二氧六环(15ml)和2m na2co3水溶液(5.0ml,10.0mmol)的混合物中的中间体68(250mg,0.59mmol)、嘧啶-5-硼酸频哪醇酯(304mg,1.48mmol)和pd(dppf)cl2·
dcm(48.2mg,0.06mmol)在130℃下加热30分钟。将rm在etoac(20ml)与水(20ml)之间进行配分,并且将分离的水层用etoac(20ml)进行萃取。将合并的有机层用盐水(20ml)进行洗涤,干燥(mgso4),在真空中浓缩,并且将剩余物通过柱色谱进行纯化以得到作为灰色固体的标题化合物(174mg,78.5%)。lcms(es
+
):376.0[mh]
+
。rt 5.96分钟,100%纯度。
[0520]
参照例3
[0521]
4-甲基-3-(3-吡啶基)-n-[4-(三氟甲氧基)苯基]苯甲酰胺
[0522][0523]
将中间体69(2.40g,5.70mmol)、吡啶-3-基硼酸(1.05g,8.55mmol)、na2co3(1.81g,17.1mmol)和pd(pph3)4(400mg,570μmol)溶解在etoh/dme/水(13.5ml,1.5∶10∶2)中。使用微波反应器将反应加热至130℃持续30分钟。将溶剂在真空中除去并且将剩余物通过柱色谱和rp hplc进行纯化以得到作为白色固体的标题化合物(320mg,15.1%
$
)。lcms(es
+
):373.1[mh]
+
。uplc:rt 4.93分钟,99.1%纯度。
[0524]
参照例4
[0525]
5-(3-吡啶基)-n-[4-(三氟甲氧基)苯基]吡啶-3-甲酰胺
[0526][0527]
类似于一般的酰胺化步骤c,使用中间体71和4-(三氟甲氧基)苯胺以得到作为白色固体的标题化合物(104mg,58.1%
$
)来合成参照例4。lcms(es
+
):360.1[mh]
+
。uplc:rt 4.44分钟,100%纯度。
[0528]
参照例5
[0529]
2-氧代-1-(嘧啶-5-基)-n-[4-(三氟甲氧基)苯基]-1,2-二氢吡啶-3-甲酰胺
[0530][0531]
向meoh(10ml)中的中间体58(483mg,1.62mmol)、(嘧啶-5-基)硼酸(206mg,1.62mmol)和cu(otf)2(604mg,1.62mmol)添加吡啶(0.27ml,2.56mmol),然后在具有泡沫计数器的密封容器中将混合物在25℃下搅拌16小时。将沉淀物滤出并且将滤液在真空下浓缩。将剩余物在etoac(20ml)中溶解,用氨水溶液(2
×
50ml)进行洗涤,然后将有机层经na2so4干燥,在真空下浓缩并且通过hplc进行纯化以得到作为米色固体的标题化合物(33.0mg,5.2%)。lcms(es
+
):377.0[mh]
+
。rt 2.67分钟,99.6%纯度。
[0532]
高分辨率质谱数据
[0533]
[0534]
[0535][0536]
生物数据
[0537]
ba/f3 celltiter-glo测定
[0538]
细胞滴度-glo发光细胞生存力测定是基于存在的atp的量化的确定培养物中活细胞数的同质性方法。简言之,修饰il-3依赖性ba/f3细胞以表达bcr-abl。转化激酶的活性克
服了il3对细胞增殖和存活的依赖性。特异性抑制激酶活性的测试化合物导致程序性细胞死亡,这可以通过添加细胞滴度-glo试剂来测量。在该测定中,表达bcr-abl的ba/f3细胞(高级细胞动力学)或亲本ba/f3(对照)细胞在含有10%fbs、1
×
glutamax和750ng/ml嘌呤霉素的rpmi 1640中以5
×
104/ml制备。将测试化合物使用tecan d300e以10μm的最高最终测定浓度分配到384孔板中,剂量归一化为50μl体积中的0.1%dmso。向准备的384孔板的每个孔添加50μl细胞,并将板以1000rpm旋转1分钟,然后在37℃、5%co2下孵育48小时。在48小时之后,向板的每个孔添加15μl细胞滴度glo试剂。在室温孵育60分钟之后,在pherastar fs读取仪上读取发光。
[0539]
在ba/f3 celltiter-glo测定中对本发明的示例性化合物进行测试并且将ic
50
数据示于表9中。本发明的所有示例性化合物的ic
50
值均为1000nm或更小。该数据表明本发明的化合物可以抑制c-abl。
[0540]
参照例5的ic
50
值》10μm并因此对c-abl无活性。不希望受到理论束缚,参照例5中的吡啶酮c=o键可能与酰胺基在空间上冲突,从而诱导酰胺-吡啶酮键中的不利的扭曲,使得两个部分不再共平面。这可能破坏吡啶酮环与c-abl的tyr454剩余物之间的边对面π-堆叠相互作用。此外,吡啶酮c=o键可以通过与酰胺基的nh的分子内氢键来形成6元环,这阻止在c-abl的活性位点内,nh与水分子形成潜在的关键的氢键,并且甚至可以替代所述水分子。本发明中的化合物的吡啶酮区域异构体没有这些缺点,并因此表现出提高得多的对c-abl的抑制。
[0541]
表9:bcr-abi ic
50
(a:1000nm至151nm,b:150nm至31nm,c:≤30nm)
[0542]
实施例ic
50
实施例ic
50
实施例ic
50
参照例1c29b62a参照例2c30c63c参照例3b31b64b参照例4b32a65c参照例5>10μm33b66b1a34b67c2a35c68c3b36b69c4a37c70b5b38c71c6a39b72c7a40b73c8a41a74a9b42c75b10b43a76a11c44c77a12b45a78a13b46c79a14a47c80a
15b48b81b16a49b82b17a50b83b18c51c84a19a52b85a20b53c86b21a54c87c22a55b88b23a56c89c24c57b90a25b58b91b26a59b92a27b60b93a28a61c94b
[0543]
大鼠中的药代动力学谱和脑外显率的确定
[0544]
将300至350g雄性sprague dawley大鼠(charles river,uk)在12小时光/暗循环下分组圈养,n=3,可随意获得食物和水。在给药前一天17:00取出所有食物。在给药当天,将动物称重,标记尾巴并通过经口管饲给药体积为5ml/kg中的3mg/kg化合物。在给药之后30分钟、1小时和4小时通过腹膜内施用戊巴比妥(pentobarbital)处死动物。通过心脏穿刺抽取死后血液,并在在冰上短暂储存在k2 edta血液管中,然后在4℃下以14,000g旋转4分钟。将血浆取出至96孔板中,置于干冰上并储存在-80℃下。将脑快速解剖并置于干冰上,然后储存在-80℃下。如下所述进行血浆和脑样品的生物分析。用来自工业标准文献
i,ii
的引导来准备方法。
[0545]
血浆生物分析
[0546]
使用10mm dmso储备液(stock)在稀释剂(mecn∶水,1;1)中在10至100,0000ng/ml的范围内制备测试化合物的添加溶液(spiking solution)。通过将2.5μl校准添加溶液添加到25μl对照血浆中以在1至10000ng/ml的范围内的已知浓度制备在对照雄性sprague-dawley大鼠血浆中的校准线。将实验样品解冻至室温,并使用蛋白质沉淀(在室温用含有25ng/ml甲磺丁脲作为内标的400μl mecn搅拌至少5分钟)沿着校准线萃取25μl等分样品。通过在4℃下以4000rpm离心5分钟将蛋白质沉淀物与萃取的测试化合物分离。将所得上清液用相关稀释剂(例如在水中的0.1%甲酸或1∶1meoh∶水)以1∶2的比率稀释。通过uplc-ms/ms在api6500 qtrap或waters 30 tqs质谱仪上使用特定于测试化合物的先前优化的分析mrm(多反应监测)方法对样品进行分析。在对照校准线的两个复制品分析样品之后,确定分离样品中测试化合物的浓度,在使用适当回归和加权的样品组之前和之后注射。校准线仅包含20%预期测试浓度范围内的样品,并且任何超出校准线限制的样品均将被视为低于或高于量化限制(lloq/aloq)。
[0547]
脑生物分析
[0548]
使用10mm dmso储备液在稀释剂(1∶1 mecn∶水)中在10至100,000ng/ml的范围内制备测试化合物的添加溶液。通过将2.5μl校准添加溶液添加到25μl对照匀浆中,以在3至
30000ng/g的范围内的已知浓度制备在对照雄性sprague-dawley大鼠脑匀浆中的校准线。为了制备对照和实验脑匀浆,将脑解冻、称重并以每克脑2ml的比率添加一定体积的稀释剂(水)。使用precellys evolution和ck14 7ml小陶瓷珠匀浆管通过珠-搅拌器匀浆进行脑的匀浆。使用蛋白质沉淀(在室温下用含有25ng/ml甲磺丁脲作为内标的400μl mecn搅拌至少5分钟)沿着校准线萃取25μl实验样品的等分样品。通过在4℃下以4000rpm离心5分钟将蛋白质沉淀物与萃取的测试化合物分离。将所得上清液用相关稀释剂(例如在水中的0.1%甲酸或1∶1 meoh∶水)以1∶2的比率稀释。通过uplc-ms/ms在api6500q trap或waters tqs质谱仪上使用对测试化合物具有特异性的先前优化的分析mrm(多反应监测)方法分析样品。在对照校准线的两个复制品分析样品之后,确定分离样品中测试化合物的浓度,在使用适当回归和加权的样品组之前和之后注射。校准线仅包含20%预期测试浓度范围内的样品,并且任何超出校准线限值的样品均将被视为低于或高于量化限制(lloq/aloq)。
[0549]
脑与血浆(b∶p)比率的确定
[0550]
通过对于各个时间点,将脑中的浓度除以血浆中的浓度计算总的脑与血浆(b∶p)比率。通过在某个时间点将这些脑与血浆比率求平均值计算平均的脑与血浆比率。表10示出了对于本发明和参照例的化合物的脑与血浆(b∶p)比率。与参照例相比,本发明的实施例具有提高得多的脑与血浆(b∶p)比率。因此,本发明的化合物特别地可用于对其中血脑屏障渗透是重要的某些疾病和病症进行的治疗中。注意,脑血屏障渗透是不可预测的并且是根据经验确定的。克服与将治疗剂递送至脑的特定区域相关的挑战对大多数脑障碍的治疗提出了主要挑战。
[0551]
表10:大鼠中的脑与血浆比率
[0552]
[0553][0554]
体内生物利用度、清除率和半衰期的确定
[0555]
通过使用phoenix winnonlin 64软件(设置8.0)的非房室分析来计算药代动力学参数,例如生物利用度(%f)、清除率(cl)、半衰期(t1/2)和分布的体积。由p.o.给药的大鼠计算生物利用度,而由i.v.给药的大鼠计算清除率和半衰期。简言之,将体内血浆浓度、时间点和剂量值以兼容格式导入软件中。将对于各动物的血浆浓度对时间绘图,并且识别并选择消除相。使用线性梯形的线性内插来计算用于各个图的曲线下的面积,由该线性内插可以随后确定药代动力学参数。表11示出了用于本发明和参照例的化合物的药代动力学参数。与参照例相比,本发明的化合物具有提高得多的清除率(cl)和半衰期(t1/2)。
[0556]
表11:大鼠pk谱
[0557]
[0558][0559]
动力学水溶解度测定
[0560]
使用100%dmso中的测试化合物的10mm原液溶液,将测试化合物以250μm的最终浓度掺加到0.05m磷酸钾缓冲剂(ph 7.4)中。使缓冲剂中的样品(96孔板中的n=2水溶液样品)在室温下在轨道振荡器上平衡30分钟(300rpm)以诱导测试化合物的沉淀。通过视觉检查确定每个样品的外观并进行记录(透明、浑浊、观察到的沉淀物等)。使用multiscreen hts溶解度过滤板(millipore)过滤磷酸盐水溶液缓冲剂样品,并且通过lc-uv沿着在50∶50乙腈∶水中以5μm、25μm、100μm和250μm制备的测试化合物的校准标准来对滤液进行分析。通过将每个重复的uv吸收峰面积与校准标准的uv吸收峰面积进行比较来确定磷酸盐缓冲剂滤液中的化合物的浓度。表12示出了用于本发明和参照例的化合物的动力学水溶解度值。与参照例相比,本发明的化合物具有提高得多的溶解度。
[0561]
表12:动力学水溶解度
[0562]
[0563][0564]
herg测定
[0565]
入ether
‑á‑
go-go相关基因(human ether
‑á‑
go-go related gene,herg)钾通道(kv11.1)有助于人心脏动作电位复极化。herg通道的抑制可以延长人心脏动作电位,从而导致qtc延长和潜在致命的心律失常(例如,尖端扭转型室性心动过速(torsade de pointes))。
[0566]
使用稳定表达编码herg通道的人ether
‑á‑
go-go相关基因的中国仓鼠卵巢(chinese hamster ovary,cho)细胞系,在qpatch 48吉咖封口(gigaseal)自动化膜片钳平台上针对herg通道筛选测试样品。在常规的全细胞配置中制作所有记录并使用标准单孔芯片(rchip 1.5mω至4mω)在室温(约21℃)下进行所有记录。将串联电阻(4mω至15mω)补偿》80%。使用以0.1hz的刺激频率施加的工业标准“+
40/-40”电压方案来从-90mv的保持电位中引发电流。在实现全细胞配置时,将载剂(外部记录溶液中的0.1%dmso v/v)在两个大丸剂添加物中施加至各细胞,其中每个添加物之间的记录周期为2分钟。在载体期之后,施加从0.003μm至10μm的八个增加浓度的测试样品作为每测试浓度的单个大丸剂添加物,并且在2分钟记录周期期间测量对herg尾部电流幅度的影响。针对相同的细胞,使用各个浓度的累积单个样品添加物构建每个八点浓度-响应曲线。该研究中使用的阳性对照为盐酸维拉帕米(tocris,目录#0654,分批#5a/61673)并且被制备成100%dmso中的10mm的原液浓度并且保持为冷冻等分试样。
[0567]
数据分析:使用qpatch软件选择所有通过qc参数(即,》200pa外向电流、膜电阻和衰减)的细胞作为“通过的qc”,然后在由光标位置限定的各个浓度施加期结束时,将其计算对于最后三次扫描的平均峰电流。对于各个测试浓度施加期计算百分比抑制作为相对于在对照(即,载剂)期结束时测量的光标值的平均光标值(峰电流或电荷)的减少。使用来自各细胞的百分比抑制值以构建浓度-响应曲线,所述浓度-响应曲线采用自由hill斜率因子和分别用以非常低的浓度和非常高的浓度固定的0%和100%抑制水平的四个参数逻辑拟合。
current regulatory guidances.j anal bioanal techniques s4:001.doi:10.4172/2155-9872.s4-001.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1