2-氯-1-(2-氯噻唑-5-基)乙酮的制备的制作方法
2-氯-1-(2-氯噻唑-5-基)乙酮的制备
发明领域
1.本发明涉及一种制备2-氯-1-(2-氯噻唑-5-基)乙酮的方法。
2.发明背景
3.2-氯-1-(2-氯噻唑-5-基)乙酮是特别由wo 2014/164084 a1已知的吡啶鎓化合物合成中的有价值的中间体。这些化合物显示了优异的杀虫特性。
4.wo 2018/197541 a1和wo/202654 a1公开了这些吡啶鎓化合物的合成路线,其包括使氯-(2-氯噻唑-5-基)镁物质与2-氯-n-甲氧基-n-甲基-乙酰胺反应形成2-氯-1-(2-氯噻唑-5-基)乙酮。
5.t.chalopin等,second generation of thiazolylmannosides,fimh antagonists for e.coli-induced crohn’s disease,org.biomol.chem.,2016,14,3913-3925描述了由硫脲合成1-(2-氯噻唑-5-基)乙酮。
6.然而,现有技术的方法具有几个缺点,例如高的流出物负载和金属盐负载、低产率、低选择性和使用有毒溶剂。
7.因此,本发明的目的是提供一种经济的制备2-氯-1-(2-氯噻唑-5-基)乙酮的方法,该方法以高的总收率,即≥85%或≥80%,高选择性地提供2-氯-1-(2-氯噻唑-5-基)乙酮,该方法可以容易地控制。
8.发明概述
9.令人惊奇的是,已经发现,通过以特定的加入速率加入氯丙酮,然后进行酸处理,桑德迈尔(sandmeyer)反应和氯化,1,3-双(二甲氨基亚甲基)硫脲和氯丙酮的受控环化导致在合理的工艺时间内以高产率和高选择性形成2-氯-1-(2-氯噻唑-5-基)乙酮。
10.因此,在一个方面,本发明涉及制备2-氯-1-(2-氯噻唑-5-基)乙酮的方法,其至少包括以下步骤:
11.a)使1,3-双(二甲氨基亚甲基)硫脲与氯丙酮环化以形成n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒,
12.b)使(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒经受至少一种酸处理以形成1-(2-氨基噻唑-5-基)乙酮,
13.c)使1-(2-氨基噻唑-5-基)乙酮进行桑德迈尔反应以形成1-(2-氯噻唑-5-基)乙酮,和
14.d)使1-(2-氯噻唑-5-基)乙酮氯化以形成2-氯-1-(2-氯噻唑-5-基)乙酮,其中在步骤a)中,以基于氯丙酮的总体积计为≥2.0体积%/分钟至≤8.0体积%/分钟的速率加入氯丙酮。
15.详述
16.在描述本技术所要求保护的发明的组合物和配制剂之前,应理解的是,本发明不限于所述的特定组合物和配制剂,因为该组合物和配制剂通常可以改变。还应理解的是,本文所用的术语并非意欲是限制性的,因为本技术要求保护的发明的范围仅由所附权利要求书限定。
17.如果下文将组定义为至少包括一定数量的实施方案,则这意味着还包括优选仅由这些实施方案组成的组。此外,说明书和权利要求中的术语“第一”、“第二”、“第三”或“a”、“b”、“c”等用于区分类似的要素,而不一定用于描述顺序或时间次序。应理解的是,如此使用的术语在适当的情况下为可互换的,并且本文描述的本技术所要求保护的发明的实施方案能够以不同于本文描述或示出的顺序操作。在术语“第一”、“第二”、“第三”或“(a)”、“(b)”和“(c)”或“(a)”、“(b)”、“(c)”、“(d)”、“i”、“ii”等涉及方法或用途或测定的步骤的情况下,步骤之间没有时间或时间间隔连贯性,即步骤可以同时进行或在这些步骤之间可以存在数秒、数分钟、数小时、数天、数周、数月或甚至数年的时间间隔,除非在如上文或下文所述的本技术中另有说明。
18.此外,在整个说明书中定义的范围也包括端值,即1-10的范围意味着1和10都包括在该范围内。为了避免疑惑,申请人应被授予根据适用法律的任何等同方案。
19.在以下段落中,更详细地定义了本技术所要求保护的发明的不同方面。如此定义的每个方面可与任何其他一个或多个方面组合,除非明确地相反说明。特别地,任何被指示为优选或有利的特征可与任何其他被指示为优选或有利的一个或多个特征组合。
20.在整个说明书中对“一个实施方案”或“实施方案”的引用意味着就在本技术所要求保护的发明的至少一个实施方案中包括该实施方案所述的特定特征、结构或特性。因此,在本说明书中的各个地方出现的短语“在一个实施方案中”或“在实施方案中”不一定全部指同一实施方案,而是可以指同一实施方案。此外,下文中使用的术语“优选”、“更优选”、“甚至更优选”、“最优选”和“特别是”或类似术语与任选特征一起使用,而不限制替代可能性。因此,由这些术语引入的特征是任选的特征并不意欲以任何方式限制权利要求的范围。
21.此外,如本领域技术人员由本公开内容知悉的那样,在一个或多个实施方案中,可以以任何合适的方式组合特定特征、结构或特性。此外,尽管本文所述的一些实施方案包括一些但不包括在其他实施方案中的其他特征,但是不同实施方案的特征的组合意味着处于本技术所要求保护的发明范围内,并且形成不同的实施方案,如本领域技术人员理解的那样。例如,在所附权利要求书中,任何要求保护的实施方案可以以任何组合使用。
22.此外,应注意的是表示一个特征或要素可能存在一次或超过一次的术语“至少一个”、“一个或多个”或类似的表述通常在介绍各自的特征或要素时仅使用一次。在下文中,在大多数情况下,当提及各自的特征或要素时,表述“至少一个”或“一个或多个”将不再重复,尽管存在各自的特征或要素可能存在一次或超过一次的事实。
23.缩合
24.本发明方法使用1,3-双(二甲氨基亚甲基)硫脲作为起始材料。
25.在优选的实施方案中,1,3-双(二甲氨基亚甲基)硫脲通过硫脲与二甲基甲酰胺二烷基缩醛,更优选二甲基甲酰胺二甲基缩醛的缩合来获得。
26.在一个优选的实施方案中,缩合在≥40℃至≤70℃的温度下进行,更优选在≥50℃至≤65℃的温度下进行。
27.在优选的实施方案中,缩合在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行。
28.步骤a:环化
29.在本发明要求保护的方法的步骤a)中,1,3-双(二甲氨基亚甲基)硫脲和氯丙酮反
应以形成n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒,其中将氯丙酮以基于氯丙酮的总体积计为≥2.0体积%/分钟至≤8.0体积%/分钟的速率加入1,3-双(二甲氨基亚甲基)硫脲悬浮液中。
30.在优选的实施方案中,以基于氯丙酮的总体积计为≥2.0体积%/分钟至≤7.0体积%/分钟的速率,更优选以≥2.0体积%/分钟至≤6.0体积%/分钟的速率,甚至更优选以≥3.0体积%/分钟至≤6.0体积%/分钟的速率加入氯丙酮,例如,在20分钟内加入650ml氯丙酮的总体积相当于每分钟加入32.5ml,这转化为基于氯丙酮的总体积计5.0体积%/分钟的加入速率。特定的加入速率一方面允许在快速加入氯丙酮时以受控的方式进行反应,即防止反应混合物的温度的任何急剧升高,另一方面允许在缓慢加入氯丙酮时减少杂质的量。
31.在优选的实施方案中,将氯丙酮作为在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂;更优选至少一种选自一氯苯、邻二氯苯、硝基苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中的溶液加入;最优选地,至少一种溶剂是一氯苯。
32.在优选的实施方案中,步骤a)在≥40℃至≤75℃的温度下进行,更优选在≥40℃至≤70℃的温度下进行。
33.在优选的实施方案中,步骤a)在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂;更优选至少一种选自一氯苯、邻二氯苯、硝基苯甲、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行;最优选地,所述至少一种溶剂是一氯苯。
34.在优选的实施方案中,在步骤a)中,氯丙酮在至少一种溶剂中的浓度为≥100g/l至≤800g/l,更优选≥200g/l至≤700g/l,最优选≥400g/l至≤600g/l。
35.在优选的实施方案中,在步骤a)中,1,3-双(二甲氨基亚甲基)硫脲在至少一种溶剂中的浓度为≥54g/l至≤220g/l。
36.在优选的实施方案中,在步骤a)中,氯丙酮与1,3-双(二甲氨基亚甲基)硫脲的摩尔比为≥1.20:1.00至≤1.01:1.00,更优选≥1.10:1.00至≤1.01:1.00。
37.在另一个优选的实施方案中,步骤a)在不存在任何胺的情况下进行,更优选地,步骤a)在不存在任何叔胺和仲胺的情况下进行,甚至更优选地,在不存在选自三甲胺、三乙胺、甲基二乙胺、三丙胺、三异丙胺、二异丙基乙胺、二异丙胺和二丁胺的任何胺的情况下进行。
38.步骤b:水解
39.在本发明要求保护的方法的步骤b)中,n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒与至少一种酸作用以形成1-(2-氨基噻唑-5-基)乙酮。
40.在优选的实施方案中,在步骤b)中,至少一种酸选自无机酸和有机酸。优选地,无机酸选自硫酸、盐酸、磷酸、高氯酸、硝酸、亚硝酸、亚硫酸、氯酸、亚氯酸、硼酸和次氯酸。优选地,有机酸选自乳酸、乙酸、甲酸、柠檬酸、草酸、尿酸、苹果酸、甲磺酸、三氟甲磺酸、三氯甲磺酸和对甲苯磺酸。在更优选的实施方案中,在步骤b)中,所述至少一种酸是盐酸或硫酸。
41.在优选的实施方案中,在步骤b)中ph为≥1.5至≤8.0。
42.在优选的实施方案中,步骤b)在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂;更优选至少一种选自一氯苯、邻二氯苯、硝基苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行;最优选地,所述至少一种溶剂是一氯苯。
43.在优选的实施方案中,步骤b)在≥20℃至≤110℃的温度下进行,更优选在≥40℃至≤90℃的温度下进行,甚至更优选在≥50℃至≤70℃的温度下进行。
44.步骤c:桑德迈尔反应
45.在本发明要求保护的方法的步骤c)中,1-(2-氨基噻唑-5-基)乙酮经由桑德迈尔反应转化为1-(2-氯噻唑-5-基)乙酮。
46.在优选的实施方案中,步骤c)在至少一种亚硝化试剂和至少一种氯源的存在下进行,所述亚硝化试剂优选选自亚硝酸钠、亚硝酸钾和亚硝酸烷基酯,所述氯源优选选自氯化铜(ii)、四烷基氯化铵、氯化钠和盐酸。在更优选的实施方案中,所述至少一种亚硝化试剂是亚硝酸钠,所述至少一种氯源是氯化钠和/或盐酸。在优选的实施方案中,至少一种氯源与1-(2-氨基噻唑-5-基)乙酮的摩尔比为≥2.0:1.0至≤1.0:1.0。
47.在优选的实施方案中,步骤c)在至少一种催化剂的存在下进行,更优选至少一种催化剂选自铜、卤化铜(i)、硫酸铜(ii)和卤化铜(ii),甚至更优选至少一种催化剂是硫酸铜(ii)。
48.在优选的实施方案中,在步骤c)中,至少一种催化剂与1-(2-氨基噻唑-5-基)乙酮的摩尔比为≥0.01:1.0至≤0.05:1.0。
49.在另一个优选的实施方案中,步骤c)在≥-10℃至≤40℃的温度下进行,更优选在≥-10℃至≤20℃的温度下进行。
50.在优选的实施方案中,步骤c)在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂;更优选至少一种选自一氯苯、邻二氯苯、硝基苯甲、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行;最优选地,所述至少一种溶剂是一氯苯。
51.在优选的实施方案中,步骤a)、b)和c)在一锅反应中进行,更优选步骤a)、b)和c)在一锅反应中进行,其中使用一氯苯作为溶剂。
52.步骤d:氯化
53.在根据本发明要求保护的方法的步骤d)中,1-(2-氯噻唑-5-基)乙酮经由选择性氯化转化为2-氯-1-(2-氯噻唑-5-基)乙酮。
54.在优选的实施方案中,步骤d)在至少一种选自亚砜、环丁砜、乙腈、二氯甲烷、二氯乙烷、一氯苯、邻二氯苯、甲苯、碳酸二甲酯、碳酸亚乙酯、水、乙酸、甲酸、四氢呋喃、2-甲基-四氢呋喃、二甲基甲酰胺、二甲基乙酰胺、甲醇、乙醇、异丙醇、叔丁醇、正丁醇、叔戊醇和离子液体的极性溶剂中进行,更优选所述至少一种溶剂为乙腈。合适的离子液体选自1-烷基-3-甲基咪唑鎓四氟硼酸盐、1-烷基-3-甲基咪唑鎓卤化物、n-烷基吡啶鎓四氟硼酸盐和五氟磷酸盐衍生物。
55.在优选的实施方案中,步骤d)在至少一种氯化剂存在下进行,更优选至少一种氯化剂选自氯气、氰尿酰氯、1,3-二氯-5,5-二甲基乙内酰脲、n-氯琥珀酰亚胺、n-氯邻苯二甲酰亚胺、二氯异氰脲酸钠、三甲基甲硅烷基氯、草酰氯、甲磺酰氯、硫酰氯、乙酰氯、三乙基三
氯化铵、烷基咪唑鎓氯、氯化钠和fecl3。
56.在优选的实施方案中,步骤d)在至少一种酸催化剂的存在下进行,所述酸催化剂选自无机酸和有机酸。合适的无机酸包括硫酸、盐酸、磷酸、高氯酸、硝酸、亚硝酸、亚硫酸、氯酸、亚氯酸、硼酸和次氯酸。合适的有机酸包括乳酸、乙酸、甲酸、柠檬酸、草酸、尿酸、苹果酸、甲磺酸、三氟甲磺酸、三氯甲磺酸和对甲苯磺酸。
57.在优选的实施方案中,步骤d)在≥0℃至≤40℃的温度下进行,更优选在≥0℃至≤20℃的温度下进行。
58.优点
59.要求保护的本发明与以下优点中的至少一个相关联:
60.(i)从1,3-双(二甲氨基亚甲基)硫脲到1-(2-氯噻唑-5-基)乙酮的反应顺序可以在一锅中进行。
61.(ii)以高的总收率和高纯度由1,3-双(二甲氨基亚甲基)硫脲提供1-(2-氯噻唑-5-基)乙酮。
62.(iii)以高的总收率和高选择性由1,3-双(二甲氨基亚甲基)硫脲提供2-氯-1-(2-氯噻唑-5-基)乙酮。
63.(iv)本发明方法使用工艺温和的溶剂。
64.(v)本发明方法涉及低流出物负载和低金属负载。
65.(vi)本发明方法是安全的并且可以容易地控制。
66.(vii)本发明方法是经济的,因为在合理的工艺时间内加入氯丙酮。
67.下面,提供了实施方案的列表以进一步说明本公开,而不旨在将本公开限制于下面列出的具体实施方案。
68.实施方案:
69.1.一种制备2-氯-1-(2-氯噻唑-5-基)乙酮的方法,其至少包括以下步骤:
70.a)使1,3-双(二甲氨基亚甲基)硫脲与氯丙酮环化以形成n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒,
71.b)使(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒经受至少一种酸处理以形成1-(2-氨基噻唑-5-基)乙酮,
72.c)使1-(2-氨基噻唑-5-基)乙酮进行桑德迈尔反应以形成1-(2-氯噻唑-5-基)乙酮,和
73.d)使1-(2-氯噻唑-5-基)乙酮氯化以形成2-氯-1-(2-氯噻唑-5-基)乙酮,其中在步骤a)中,以基于氯丙酮的总体积计为≥2.0体积%/分钟至≤8.0体积%/分钟的速率加入氯丙酮。
74.2.根据实施方案1所述的方法,其中在步骤a)中,以基于氯丙酮的总体积计为≥2.0体积%/分钟至≤7.0体积%/分钟的速率加入氯丙酮。
75.3.根据实施方案1所述的方法,其中1,3-双(二甲氨基亚甲基)硫脲通过硫脲与二甲基甲酰胺二烷基缩醛的缩合来获得。
76.4.根据实施方案3所述的方法,其中缩合在≥40℃至≤70℃的温度下进行。
77.5.根据实施方案3或4所述的方法,其中缩合在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行。
78.6.根据实施方案1-5中任一项所述的方法,其中步骤a)在≥40℃至≤75℃的温度下进行。
79.7.根据实施方案1-6中任一项所述的方法,其中步骤a)在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行。
80.8.根据实施方案7所述的方法,其中在步骤a)中,氯丙酮在至少一种溶剂中的浓度为≥100g/l至≤800g/l。
81.9.根据实施方案1-8中任一项所述的方法,其中在步骤a)中,1,3-双(二甲氨基亚甲基)硫脲在至少一种溶剂中的浓度为≥54g/l至≤220g/l。
82.10.根据实施方案1-9中任一项所述的方法,其中在步骤a)中,氯丙酮与1,3-双(二甲氨基亚甲基)硫脲的摩尔比为≥1.20:1.00至≤1.01:1.00。
83.11.根据实施方案1-10中任一项所述的方法,其中步骤a)在不存在任何胺的情况下进行。
84.12.根据实施方案1-11中任一项所述的方法,其中在步骤b)中,至少一种酸选自无机酸和有机酸。
85.13.根据实施方案12所述的方法,其中无机酸选自硫酸、盐酸、磷酸、高氯酸、硝酸、亚硝酸、亚硫酸、氯酸、亚氯酸、硼酸和次氯酸。
86.14.根据实施方案12所述的方法,其中有机酸选自乳酸、乙酸、甲酸、柠檬酸、草酸、尿酸、苹果酸、甲磺酸、三氟甲磺酸、三氯甲磺酸和对甲苯磺酸。
87.15.根据实施方案12所述的方法,其中在步骤b)中,所述至少一种酸是盐酸或硫酸。
88.16.根据实施方案1-15中任一项所述的方法,其中在步骤b)中ph为≥1.5至≤8.0。
89.17.根据实施方案1-16中任一项所述的方法,其中步骤b)在至少一种选自一氯苯、邻二氯苯、硝基苯、二氯甲烷、二氯乙烷、氯仿、甲苯、邻二甲苯、间二甲苯和对二甲苯的溶剂中进行。
90.18.根据实施方案1-17中任一项所述的方法,其中步骤b)在≥20℃至≤110℃的温度下进行。
91.19.根据实施方案1-18中任一项所述的方法,其中步骤c)在至少一种亚硝化试剂和至少一种氯源的存在下进行。
92.20.根据实施方案19所述的方法,其中至少一种亚硝化试剂选自亚硝酸钠、亚硝酸钾和亚硝酸烷基酯。
93.21.根据实施方案19所述的方法,其中至少一种氯源选自氯化铜(ii)、四烷基氯化铵、氯化钠和盐酸。
94.22.根据实施方案19-21中任一项所述的方法,其中至少一种氯源与1-(2-氨基噻唑-5-基)乙酮的摩尔比为≥2.0:1.0至≤1.0:1.0。
95.23.根据实施方案1-22中任一项所述的方法,其中步骤c)在至少一种催化剂的存在下进行。
96.24.根据实施方案23所述的方法,其中至少一种催化剂选自铜、卤化铜(i)、硫酸铜(ii)和卤化铜(ii)。
97.25.根据实施方案23或24所述的方法,其中在步骤c)中,至少一种催化剂与1-(2-氨基噻唑-5-基)乙酮的摩尔比为≥0.01:1.0至≤0.05:1.0。
98.26.根据实施方案1-25中任一项所述的方法,其中步骤c)在≥-10℃至≤40℃的温度下进行。
99.29.根据实施方案1-26中任一项所述的方法,其中步骤d)在至少一种选自亚砜、环丁砜、乙腈、二氯甲烷、二氯乙烷、一氯苯、邻二氯苯、碳酸二甲酯、碳酸亚乙酯和离子液体的极性溶剂中进行。
100.28.根据实施方案1-27中任一项所述的方法,其中步骤d)在至少一种氯化剂存在下进行。
101.29.根据实施方案28所述的方法,其中至少一种氯化剂选自氯气、氰尿酰氯、1,3-二氯-5,5-二甲基乙内酰脲、n-氯琥珀酰亚胺、n-氯邻苯二甲酰亚胺、二氯异氰脲酸钠、三甲基甲硅烷基氯、草酰氯、甲磺酰氯、硫酰氯、乙酰氯、三乙基三氯化铵、烷基咪唑鎓氯、氯化钠和fecl3。
102.30.根据实施方案1-29中任一项所述的方法,其中步骤d)在≥0℃至≤40℃的温度下进行。
103.31.根据实施方案1-30中任一项所述的方法,其中步骤a)、b)和c)在一锅反应中进行。
实施例
104.通过以下非限制性的工作实施例详细说明本发明。
105.方法
106.表征通过联用的高效液相色谱法/质谱法(hplc/ms)、气相色谱法(gc)、nmr或熔点进行。
107.hplc方法:agilent eclipse plus c18,150mm
×
4.6mm id
×
5μm
108.梯度a=在水中的0.1%tfa,b=在乙腈中的0.1%tfa。
109.流速=1.4ml/min,柱形炉温度=30℃
110.梯度程序=10%b-100%b-5min,保持2min,3min-10%b。
111.运行时间=10minlcms方法1:c18柱(50mm
×
3.0mm
×
3μm)
112.梯度a=10mm甲酸铵水溶液,b=在乙腈中的0.1%甲酸
113.流速=1.2ml/min,柱形炉温度=40℃
114.梯度程序=在1.5min内10%b到100%b,保持1min 100%b,1min-10%b
115.运行时间:3.75min
[0116]1h-nmr:信号通过相对于四甲基硅烷的化学位移(ppm)、其多重性及其积分(给定氢原子的相对数)表征。使用下列缩写表征信号的多重性:m=多重峰,q=四重峰,t=三重峰,d=双重峰和s=单重峰。
[0117]
所用缩写为:h表示小时,min表示分钟,rt表示保留时间且环境温度表示20-25℃。
[0118]
实施例1
[0119]
(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲的制备
[0120][0121]
在氮气气氛下,在16l反应釜中加入硫脲(500g)、一氯苯(mcb)(5l)和二甲基甲酰胺二甲基乙缩醛(2.19l)。将反应物料在55-60℃下搅拌3小时。3小时后,将2.5l溶剂(甲醇和mcb)在真空下蒸馏,得到(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲,其为在mcb中的悬浮液。该悬浮液原样用于步骤b。[理论产率的99%,m/z=187amu(m+h
+
)],hplc rt:4.70
[0122]1h nmr(300mhz,cdcl3):3.17(s,6h),3.20(s,6h),8.92(s,2h)。步骤a:n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒的制备
[0123][0124]
在16l反应器中,向(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲在mcb的悬浮液中加入mcb(2l)。将反应器夹套设定为45℃,在氮气气氛下,在20分钟内计量加入氯丙酮(0.65l)在mcb(0.5升)中的溶液(在加入氯丙酮期间观察到25-30℃的放热)。在完全加入氯丙酮后,使反应物料达到55-60℃并搅拌0.5小时。hplc观察到>99%转化为n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒。反应物料在相同反应器中原样进行下一步骤。[理论产率的99%,m/z=198amu(m+h
+
)]。hplc rt:6.05,1h nmr(300mhz,cdcl3):2.28(s,3h),3.10(s,3h),3.12(s,3h),7.93(s,1h),8.28(s,1h)。步骤b:1-(2-氨基噻唑-5-基)乙酮的制备
[0125][0126]
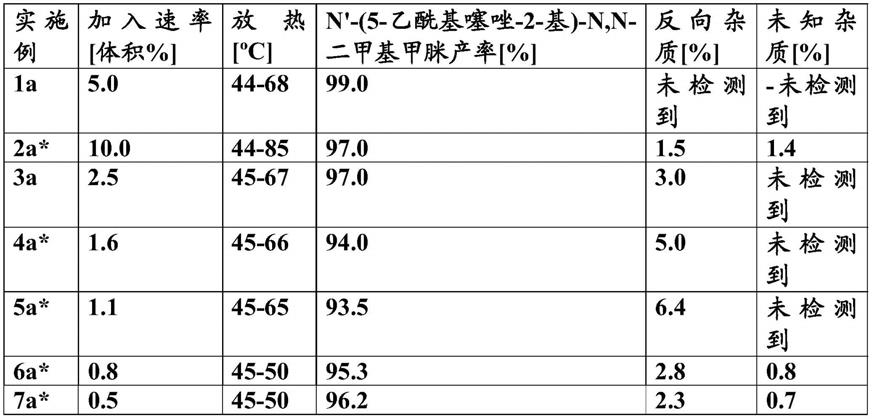
在55-60℃下在25分钟内向上述反应物料中加入hcl水溶液(35%,2.5l)(观察到5℃的放热)。在55-60℃下加入软化水(0.5l)并搅拌2小时。2小时后,hplc显示>99%转化为所需产物。停止搅拌器并在55-60℃下分离水相1。将hcl水溶液(35%,1l)加入有机相中,并在相同温度下搅拌两相混合物15-20分钟。再次分离水相2并与水相1合并。合并的水相在相同反应器中原样用于下一步骤。[理论产率的99%,m/z=143amu(m+h
+
)]。hplc rt:4.49,1h nmr(300mhz,dmsod6):2.34(s,3h),7.91(s,1h),7.99(brs,2h)。
[0127]
步骤c:1-(2-氯噻唑-5-基)乙酮的制备
[0128][0129]
向步骤b的水溶液中加入hcl水溶液(35%,1l)并将溶液冷却至-7℃。加入cuso4·
5h2o(0.16kg)和nacl(0.35kg)。在-7至3℃之间经2.5小时的时间加入nano2(0.94kg)在水(1.2l)中的溶液。在完全加入后,将反应物在10-15℃下搅拌0.5小时。加入mcb(2l)和水(2l),搅拌10分钟。分离水相,用mcb(1l)萃取。将合并的有机相用饱和nahco3水溶液(1l)和水(1l)洗涤。分离有机相,在真空下蒸馏mcb,得到1-(2-氯噻唑-5-基)乙酮(1.02kg)。[经过4步的理论产率的85%。gcms,m/z=161amu.]hplc rt:6.94,1h nmr(300mhz,cdcl3):2.58
(s,3h),8.08(s,1h)。
[0130]
步骤d:2-氯-1-(2-氯噻唑-5-基)乙酮的制备
[0131][0132]
在氮气气氛下,向装备有teflon叶片搅拌器、加料漏斗和热袋的2l 3颈烧瓶中加入1-(2-氯噻唑-5-基)乙酮(150g)和乙腈(330ml),并冷却至15-20℃。在15-20℃下于0.5小时内滴加磺酰氯(73ml)。将反应物料加热至环境温度并搅拌5小时。2-氯-1-(2-氯噻唑-5-基)乙酮的弱hcl盐从反应中沉淀出来;将悬浮液冷却至0-5℃并搅拌10分钟。将沉淀用冷乙腈(25ml)洗涤。将固体在35℃下真空干燥。在干燥过程中弱hcl盐分解,得到米色固体2-氯-1-(2-氯噻唑-5-基)乙酮[理论产率的80%,m/z=195amu(m+h
+
)]。hplc rt:7.90。1h nmr(300mhz,cdcl3):4.52(s,2h),8.21(s,1h)。
[0133]
实施例2
[0134]
(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲的制备
[0135]
在氮气气氛下,在1.6l反应釜中加入硫脲(70g)、一氯苯(mcb)(955ml)和二甲基甲酰胺二甲基乙缩醛(294ml)。将反应物料在55-60℃下搅拌3小时。3小时后,将385ml溶剂(甲醇和mcb)在真空下蒸馏,得到(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲,其为在mcb中的悬浮液。该悬浮液原样用于步骤b。[理论产率的99%,m/z=187amu(m+h
+
)],hplc rt:4.70
[0136]1h nmr(300mhz,cdcl3):3.17(s,6h),3.20(s,6h),8.92(s,2h)。步骤a:n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒的制备
[0137]
在1.6l反应器中,加入(1,3e/z)-1,3-双(二甲氨基亚甲基)硫脲在mcb中的悬浮液。将反应器夹套设定为55℃,施加200-250毫巴真空,在2-3小时内使用汲取管计量加入氯丙酮(86.8ml)在mcb(70ml)中的溶液(在加入氯丙酮期间观察到3-5℃的放热)。在完全加入氯丙酮后,使反应物料在60℃下搅拌0.5小时。hplc观察到>99%转化为n'-(5-乙酰基噻唑-2-基)-n,n-二甲基甲脒。反应物料在相同反应器中原样进行下一步骤。[理论产率的97%,m/z=198amu(m+h
+
)]。hplc rt:6.05,1h nmr(300mhz,cdcl3):2.28(s,3h),3.10(s,3h),3.12(s,3h),7.93(s,1h),8.28(s,1h)。步骤b:1-(2-氨基噻唑-5-基)乙酮的制备
[0138]
在55-60℃下在40分钟内向上述反应物料中加入hcl水溶液(35%,407ml)(观察到5℃的放热)。在55-60℃下加入软化水(70ml)并搅拌2小时。2小时后,hplc显示>99%转化为所需产物。停止搅拌器并在55-60℃下分离水相1。将hcl水溶液(35%,166ml)加入有机相中,并在相同温度下搅拌该两相混合物15-20分钟。再次分离水相2并与水相1合并。合并的水相在相同反应器中原样用于下一步骤。[理论产率的98%,m/z=143amu(m+h
+
)]。hplc rt:4.49,1h nmr(300mhz,dmsod6):2.34(s,3h),7.91(s,1h),7.99(brs,2h)。
[0139]
步骤d:2-氯-1-(2-氯噻唑-5-基)乙酮的制备
[0140]
方法-1:
[0141]
在氮气气氛下,向装备有玻璃叶片搅拌器和热袋的1l夹套反应器中加入1-(2-氯噻唑-5-基)乙酮在一氯苯(180g)中的55.5重量%溶液,用一氯苯(72ml)和乙腈(300ml)冲洗烧瓶,并冷却至15-20℃。在15-20℃下于2-3小时内吹扫氯气(90gm)。将反应物料在15-20℃下搅拌3小时。2-氯-1-(2-氯噻唑-5-基)乙酮的弱hcl盐从反应中沉淀出来。加入一氯苯
(200ml)并将悬浮液回流40分钟。蒸除溶剂(mcb:can,226ml)。向残留水(加入102ml),然后5%nahco3(90ml)。分离有机相并再用水(100ml)洗涤。真空蒸发有机相,得到2-氯-1-(2-氯噻唑-5-基)乙酮,为24%mcb溶液。[理论产率的80%,m/z=195amu(m+h
+
)]。hplc rt:7.90。1h nmr(300mhz,cdcl3):4.52(s,2h),8.21(s,1h)。
[0142]
方法-2:
[0143]
向装备有玻璃叶片搅拌器和热袋的1l夹套反应器中加入1-(2-氯噻唑-5-基)乙酮(92g)、甲苯(92g)和乙腈(368g)。在氮气气氛下,室温。在25-30℃下在2-3小时内吹扫氯气(45gm)。在室温下搅拌反应物料3小时。将反应物料冷却至0-5℃并搅拌30分钟。将从溶液中沉淀出的固体2-氯-1-(2-氯噻唑-5-基)乙酮过滤并真空干燥。[理论产率的73%,m/z=195amu(m+h
+
)]。hplc rt:7.90。1h nmr(300mhz,cdcl3):4.52(s,2h),8.21(s,1h)。
[0144]
2-氯-1-(2-氯噻唑-5-基)乙酮的结晶:
[0145]
将粗物质(571g,84.52%)溶解在ipa(856ml)中,并在60℃下加热,得到澄清溶液,搅拌30分钟,逐渐冷却至0℃,搅拌30分钟。过滤固体并真空吸干,得到2-氯-1-(2-氯噻唑-5-基)乙酮(443g,98.9重量%,理论产率的92%),为米色固体。
[0146]
发现氯丙酮向1,3-双(二甲氨基亚甲基)硫脲中的加入速率是关键的。一方面,如果氯丙酮加入太快,反应不再能控制,并且超过70℃的温度。另一方面,如果氯丙酮加入太慢,1,3-双(二甲氨基亚甲基)硫脲的产率降低,并且形成其他副产物,其对2-氯-1-(2-氯噻唑-5-基)乙酮的合成的总产率产生负面影响。
[0147]
下表1给出了不同加入速率的概述。所有实施例均按照实施例1a进行。然而,在每种情况下,改变了氯丙酮的加入速率。检测出未知杂质和反向杂质n,n-二甲基-n'-(4-甲基噻唑-2-基)甲脒。
[0148]
表1
[0149][0150]
*不在本发明范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1