α-烯烃的非均相异构化的方法与流程
1.本发明涉及通过使用包含硅-铝混合氧化物的非均相催化剂作为催化剂以连续固定床操作模式将α-烯烃异构化成相应的内烯烃的新方法。
背景技术:
2.α-烯烃具有各种各样的最终用途,主要取决于链长而定。有某些应用,例如制纸施胶剂和钻井液,其另外需要使用内烯烃。内烯烃可通过异构化方法由它们相应的α-烯烃制备。已知几种催化剂用于将α-烯烃异构化成内烯烃。活性催化剂可达到热力学异构化极限,从而得到具有平衡分布的双键异构体的内烯烃。
3.所生成的且随后被纯化的异构体的用途可以是双键的环氧化,产生环氧化物,该环氧化物可以在第二步中聚合成具有不同链长的聚酯,用于在各种应用和配制剂(如润滑剂)中作为基础油或粘度改性剂。由这些异构体产生的环氧化物的另一种应用是形成二酯,该二酯也可在多种应用和配制剂中用作低粘度基础流体。
4.已知多种催化剂用于烯烃类化合物的双键异构化。这些主要是沸石和分子筛以及树脂型酸性体系(us 5,849,974)。然而,这些催化剂中有许多会产生大量的聚合物和/或骨架异构化产物;即支化的烯烃或二聚体或低聚物。对于一些应用而言,希望将支化产物限制在尽可能少的量。因此,对于一些应用而言,希望使用对双键异构化有选择性而不会使骨架结构异构化或形成二聚体或更高级低聚物的催化剂。二聚体和更高级低聚物的形成是酸性催化剂体系的副反应。为了避免这些副反应,使用碱性催化剂或掺杂有碱金属或碱土金属的催化剂。
5.us 6,281,404 b1涉及在具有特定孔几何形状的分子筛的存在下使烯烃类进料化合物双键异构化的方法。这样的催化剂被描述为是含二氧化硅的铝磷酸盐分子筛,其通常被称为sapo。在工作实施例中,使用sapo-39将1-戊烯在400℉(204℃)的温度下异构化。实现的转化率为最高至65重量%,非常低。
6.wo 2005/031066涉及在70℃至140℃的操作温度下,在固体酸二氧化硅-氧化铝催化剂的存在下,将c16和/或c18α-烯烃的组合物转化成内烯烃的方法。据报道,异构化的内烯烃混合物含有少于25重量%的α-烯烃和相应的在6-8位具有双键的内烯烃。在工作实施例中,使用x-600(criterion catalyst公司的产品)将1-十六碳烯在70℃、100℃和120℃的温度下异构化。这种方法的一个缺点是在异构化之前需要将所述催化剂在高温(约350℃)下活化(参见例如实施例1)。
7.wo 2009/085886涉及一种在sapo催化剂的存在下将α-烯烃异构化成内烯烃的方法。然而,所述催化剂的活性是低的(whsv《0.7)。
8.us 8,575,081 b2涉及制备基于二酯的润滑剂组合物的方法。使用烯烃作为起始材料,该烯烃可以是α-烯烃或具有内部双键的异构化烯烃。这样的异构化被描述为是使用诸如结晶铝硅酸盐和铝磷酸盐的催化剂在催化情况下进行的。然而,其中没有公开任何具体的使用根据本发明的催化剂的异构化。
9.us 8,980,784 b2涉及硅-铝混合氧化物和它们作为催化剂用于气相解离甲基叔丁基醚(mtbe)的用途。其中没有公开α-烯烃的异构化。
10.us 9,371,255涉及混合氧化物组合物,其作为催化剂用于裂解烷基叔烷基醚或叔醇的用途,并涉及用于将烷基叔烷基醚或叔醇裂解成异构烯烃和醇或水的方法。然而,没有公开α-烯烃的异构化。
11.通常,所述一种或多种用于制备异构化烯烃的方法可以使用能够提供具有希望特征的烯烃产物的反应条件进行。为了形成希望的烯烃产物而可以使用和变化的异构化反应条件尤其包括反应温度、重时空速、反应压力、烯烃原料转化成异构化烯烃的转化率、反应器流出物中发现的骨架异构化烯烃的量,以及是否存在溶剂或稀释剂。
12.已知的异构化催化剂和各自方法具有以下缺点中的至少一个:
13.(a)α-烯烃转化成内烯烃的转化率低而使得方法不具有经济优势和成本效益。
14.(b)内烯烃在碳链上的分布变化。这意味着不能恒定地制备具有一致品质的,即异构化产物分布均一的获得产物。
15.(c)形成不希望的副产物,例如产物中高浓度的二聚体,其必须在随后的清洁步骤中被除去。
16.(d)催化剂的操作寿命短。
17.(e)需要高温处理或其它特定过程来活化催化剂。
18.(f)催化剂的再循环经常是不可能的,或者提供了显著不同的产物混合物。
19.(g)催化剂需要频繁的再活化(例如在空气中)。
技术实现要素:
20.因此,本发明的目的是提供一种在催化剂的存在下使α-烯烃异构化的方法,借助于该方法可以最小化或完全避免上述缺点。
21.现在已经令人惊奇地发现,通过使用具有中等或低酸度的催化剂,例如在本技术中描述的包含硅-铝混合氧化物的非均相催化剂作为催化剂,将α-烯烃异构化的方法实现了所述技术目的。这样的方法在双键异构化方面提供了有希望的结果,其中诸如支化和低聚的副反应被最小化,并且所述方法可以以间歇或连续固定床的操作模式运行,优选以连续固定床操作模式运行。这种方法也可以在连续淤浆反应器中运行。
22.本发明的一个实施方案涉及通过使用硅-铝混合氧化物作为催化剂将α-烯烃转化成异构化产物的方法。所述方法优选以连续固定床操作模式进行。
23.所述异构化产物含有内部不饱和键(即它们是内烯烃)并且只含有少量的二聚体。优选地,α-烯烃转化成异构化产物的转化率为95%或更高。在一种或多种所述异构化产物中二聚体的量优选为5摩尔%或更低。
24.在本发明中用作催化剂的混合氧化物以如在de 198 47 161 a1或ep 0850 876 a1中描述的火焰水解的方式制备。
25.在这种所谓的“共气相法(co-fumed process)”中,将挥发性硅和铝化合物,通常是四氯化硅和三氯化铝,注射到氢气和氧气或空气的爆炸性气体火焰中。所述挥发性硅和铝化合物被在爆炸性气体火焰中形成的水水解,从而形成混合氧化物。
26.上述文献中还公开的一种替代方案是掺杂方法。其中,将气溶胶进料到氢气/氧气
火焰中,其中氧化物,例如如在本发明情况下的氧化硅,是由挥发性化合物,例如四氯化硅,通过火焰水解获得的,所述气溶胶含有待掺杂元素(例如铝)的盐,并因此形成相应的混合氧化物。通过火焰水解制备的硅-铝混合氧化物主要或完全是无定形的。
27.通过上述方法制备的硅-铝混合氧化物的特征在于纯度高,并且其具有以下组成:
28.(a)75至99.99重量%,优选85至99.99重量%,更优选95至99.9重量%,和甚至更优选97至99.9重量%的氧化硅(作为sio2形式计算);
29.(b)0.01至25重量%,优选0.01至15重量%,更优选0.1至5重量%,和甚至更优选0.1至3重量%的氧化铝(作为al2o3形式计算)。
30.在另一个实施方案中,所述硅-铝混合氧化物确实另外含有碱金属或碱土金属氧化物,优选其量为基于所述硅-铝混合氧化物的总组成计的最高至1重量%。
31.为了引入所述碱金属或碱土金属,可用碱金属或碱土金属氢氧化物的水溶液处理通过火焰水解制备的混合氧化物。这例如可以通过用碱金属和/或碱土金属盐溶液浸渍通过火焰水解制备的混合氧化物来实现。随后,将所得的混合氧化物用水洗涤,在100至150℃下干燥并在300至600℃下,优选在450至550℃下煅烧。
32.所述硅-铝混合氧化物还可含有痕量的碱金属或碱土金属,它们在本发明的上下文中不被考虑。
33.本发明的另一个实施方案涉及用酸性含水磷源进一步处理的硅-铝混合氧化物。使用的磷源可以是磷酸、膦酸、次膦酸、多磷酸或磷酸二氢盐,优选磷酸。所述处理通过以下方式进行:将所述混合氧化物组合物悬浮在水中并将这种悬浮液与所述磷源混合,使得建立0至6,优选1至2.5,和更优选2至2.5的ph。随后,将经处理的催化剂用水洗涤,在100至150℃下干燥并在300至600℃下,优选在450至550℃下煅烧。
34.在另一个实施方案中,所述硅-铝混合氧化物组合物尤其主要(意思是《70%)或完全以聚集的初级粒子的形式存在。
35.根据本发明的硅-铝混合氧化物组合物的特征尤其在于在靠近表面的层中的初级粒子的重量比(al2o3/sio2)
表面
低于整体初级粒子的重量比(al2o3/sio2)
总体
。术语“靠近表面的层”意思是指从表面直到5nm深度的区域。所述重量比的差意味着氧化铝在表面处的浓度低于在整体组合物中的浓度。整体初级粒子也包括在靠近表面的层中的二氧化硅和氧化铝的部分。
36.在另一个实施方案中,所述硅-铝混合氧化物组合物主要或完全以聚集的初级粒子的形式存在,其特征在于
37.(a)在总体初级粒子中的重量比(al2o3/sio2)
总体
为0.002至0.05,优选0.003至0.015,且更优选0.005至0.01;和
38.(b)在靠近表面的层中的初级粒子的重量比(al2o3/sio2)
表面
低于在整体初级粒子中的重量比。
39.为了本发明的目的,混合氧化物是混合氧化物组分氧化铝和二氧化硅在原子水平的紧密混合物,其中初级粒子也具有si-o-al键。这些初级粒子的表面大部分或完全不含孔。
40.在表面上的重量比(al2o3/sio2)
表面
可例如通过粉末的x-射线诱导光电子能谱(xps分析)测定。可通过单个初级粒子的能量色散x射线分析(tem-edx分析)获得有关表面组成
的额外的信息。
41.在总体初级粒子中的重量比(al2o3/sio2)
总体
是通过化学或物理化学方法(例如粉末的x-射线荧光分析)测定的。
42.所述硅-铝混合氧化物组合物的特征还可在于:
43.(c)bet表面积为50至250m2/g,优选100至200m2/g(根据din iso 9277(状态:2014-01)测定。
44.已经进一步发现,对于所述硅-铝混合氧化物而言,具有300-350的邻苯二甲酸二丁酯值(以“g邻苯二甲酸二丁酯(dbp)/100g混合氧化物”为单位)可能是有利的。所述dbp值是聚集体的结构的量度。低数值对应于低水平的结构,而高数值对应于高水平的结构。300至350的优选范围对应于高水平的结构。在dbp吸收中,测量在添加限定量的dbp时由dbp测量仪的旋转叶片所吸收的力,例如扭矩(以nm为单位),其与滴定相当。在此,根据本发明的硅-铝混合氧化物显示出尖锐的明显最大值,随后在特定添加dbp时下降。可例如使用得自卡尔斯鲁厄(karlsruhe)的haake的rheocord 90仪器测量所述邻苯二甲酸二丁酯吸收。为此目的,称出12g所述硅-铝混合氧化物粉末,达到0.001g精度以内,并将其引入到捏合室中,后者借助于盖子封闭,并通过在盖子中的洞以0.0667ml/s的预定计量速率计量加入邻苯二甲酸二丁酯。在每分钟125转的电机速度下操作所述捏合机。在达到扭矩最大值后,自动关闭所述捏合机和dbp的引入。由所消耗的dbp的量和所称出的粒子的量根据下式计算所述dbp吸收:dbp值(g/100g)=(dbp的消耗量(g)/粉末的重量(g))
×
100。
45.可将所述硅-铝混合氧化物组合物施加到载体上,所述载体例如金属、聚合物或陶瓷载体,所述载体关于其中要使用所述催化剂的异构化反应是惰性的。当本发明的催化剂已施加到惰性(inner)载体上时,在测定所述硅-铝混合氧化物的组成的过程中不考虑所述惰性载体的质量和组成。
46.根据本发明使用的烯烃原料选自包含10至20个碳原子,优选14至16个碳原子,和最优选14个碳原子的α-烯烃。本发明不限于包含单一组分的原料,而是也可以使用也具有不同链长的多于一种的组分的混合物。
47.在另一个实施方案中,所述烯烃原料是包含至少90重量%的单烯烃类线性α-烯烃的α-十四碳烯。
48.可商购α-烯烃的一个来源是乙烯的低聚。可商购α-烯烃产品的第二个来源是费-托(fischer-tropsch)合成料流。通过乙烯低聚制备的可用作烯烃原料的可商购正构α-烯烃产品的一个来源是德克萨斯州伍德兰兹(the woodlands,tx)的chevron phillips chemical company lp。通过乙烯低聚制备的可用作烯烃原料的可商购正构α-烯烃产品的其它来源尤其包括ineos oligomers(比利时费卢依),shell chemicals corporation(德克萨斯州休斯敦或英国伦敦),idemitsu kosan(日本东京),和mitsubishi chemical corporation(日本东京)。所制备的以及任选从费-托合成料流中分离的可商购正构α-烯烃产品的一个来源尤其包括sasol(南非约翰内斯堡)。
49.本发明的一个实施方案涉及通过使用硅-铝混合氧化物作为催化剂使α-烯烃异构化的方法,该方法包括以下步骤:
50.(a)在反应器中提供所述α-烯烃和催化剂;
51.(b)将所述反应器惰性化;
52.(c)调整在所述反应器中的压力;
53.(d)将所述反应器加热到希望的反应温度;
54.(e)搅拌反应混合物直到平均反应时间结束;
55.(f)将所述反应混合物冷却到希望的温度以进行进一步加工;
56.(g)任选地,通过适当的技术从所述反应混合物中过滤所述催化剂;
57.(h)任选地,收集经异构化的产物;和/或
58.(i)任选地,去除不希望的副产物,例如通过蒸馏、精馏、真空蒸馏、膜分离或其它合适的分离技术。
59.所述反应器可选自固定床反应器、淤浆反应器或滴流床反应器。
60.对于在固定床反应器中进行反应的变型方案,必须将用作催化剂并通过火焰水解或热解方式制备的硅-铝混合氧化物组合物在添加粘结剂的情况下经历成型工艺,如在现有技术中公知的那样,例如以颗粒、丸剂或成型体的形式,例如片剂、圆柱体、球体、挤出物或环或更特定的通过增材制造工艺(例如3d打印)获得的几何形状。合适的粘结剂是本领域已知的,例如氧化铝、陶瓷粘土、不会显著影响催化性能的胶体或无定形沸石。
61.对于所述成型工艺,将1至20重量%的硅-铝混合氧化物组合物与上文明确指出的粘结剂以及临时助剂,例如水、水溶液、水替代物(如二醇或聚二醇),和额外的固定剂(例如纤维素醚),增塑剂(例如多糖),和/或压制助剂(例如非离子型蜡分散体)一起剧烈混合。这个操作可例如在捏合机或强力混合机中进行。随后,成型工艺,例如造粒、挤出或干压,产生用于固定床反应器的成型体。在掺入之前,将所述成型体在200至700℃的温度范围内煅烧,这至少除去了所述临时助剂。
62.在一个实施方案中,在所述反应器中的压力可以在2至6巴范围内,优选在4至6巴范围内,更优选在4.5至5巴范围内。
63.在一个实施方案中,反应温度可以在130至340℃范围内,优选在150至200℃范围内,更优选在160至190℃范围内。
64.在一个实施方案中,平均反应时间可以在150至350分钟范围内,优选在200至300分钟范围内,更优选在230至260分钟范围内。
65.本发明的另一个实施方案涉及通过使用硅-铝混合氧化物作为催化剂以连续操作模式使α-烯烃异构化的方法,该方法包括以下步骤:
66.(a)在反应区中,优选在固定床反应器中,提供所述催化剂;
67.(b)将所述反应器惰性化并调整反应器的温度到希望的反应温度;
68.(c)通过控制温度、压力和重时空速(whsv),使所述α-烯烃首先经过预热区,和然后经过所述反应区,优选从底部到顶部而连续进料;
69.(d)任选地,在使用淤浆反应器的情况下,在反应出口从所述反应混合物中移除所述催化剂;
70.(e)在所述反应区后将所述反应混合物冷却到希望的温度以进行进一步加工;
71.(f)任选地,收集经异构化的产物;和/或
72.(g)任选地,去除不希望的副产物,例如通过蒸馏、精馏、真空蒸馏或膜分离。
73.所述反应器可选自活塞流型或连续搅拌型反应器(cstr)及其组合。
74.在一个实施方案中,可以使用填充有反应混合物和自由流动的粉末化催化剂的反
应器(淤浆反应器)。原料(educt)从底部进料,并且反应混合物从顶部移出,催化剂被保留,例如被过滤元件保留。所述反应区可包含内部或外部循环。
75.在另一个实施方案中,应用具有催化剂固定床的管式反应器。所述原料可从底部或顶部进料。
76.在另一个实施方案中,可以使用连续搅拌型反应器,例如具有催化剂固定床的容器(例如berty型或carberry型反应器)或具有外部循环的管式固定床反应器(例如环管反应器)。
77.在另一个实施方案中,将连续搅拌型反应器与管式固定床组合作为最终(finishing)反应器可能是有利的。
78.在一个实施方案中,所述催化剂由通过成型工艺从硅-铝混合氧化物组合物、粘结剂和临时助剂制备的成型体组成。
79.在一个实施方案中,反应温度可以在130至340℃范围内,优选在150至200℃范围内,更优选在160至190℃范围内。
80.在一个实施方案中,重时空速(whsv)在3.0至5.5h-1
范围内,优选在3.1至4.7h-1
范围内,和最优选在3.5至3.7h-1
范围内。
81.重时空速(whsv)被定义为每单位重量催化剂每小时的进料流重量。由于加料到所述反应器中的催化剂的重量不变且始终相同,因此每小时液体流量的任何变化都会改变所述whsv。时空速度的倒数是比停留时间,即在操作条件下液体与催化剂体积接触多长时间。
82.本领域普通技术人员认识到在异构化反应温度与重时空速之间存在相关性。通常,为了获得具有等同特征的异构化产物,增加重时空速将需要增加异构化反应温度。此外,本领域普通技术人员认识到,随着催化剂老化,异构化反应温度必须升高和/或重时空速必须降低以保持烯烃反应器流出物具有所需特征。
83.通常,所述反应器流出物包含异构化产物。所述异构化产物可进一步包含异构化烯烃和/或骨架异构化烯烃。离开所述反应器的烯烃可以包括非异构化烯烃、异构化烯烃和/或骨架异构化烯烃以及低聚化烯烃。低聚化烯烃包含两个或更多个所用进料的单元,即所用进料的二聚体、三聚体或更高级异构体。本文中独立地描述了可存在于烯烃反应器流出物中的非异构化烯烃、异构化烯烃和骨架异构化烯烃以及低聚化烯烃。此外,可在烯烃反应器流出物中发现的非异构化烯烃、异构化产物、异构化烯烃、骨架异构化烯烃以及低聚化烯烃的量在本文中被独立地描述并且可以以任何组合的方式用于描述本文所述的一种或多种方法的烯烃反应器流出物。
84.如上文进一步描述的方法的特征在于α-烯烃转化成相应内烯烃的转化率为90%或更高,优选95%或更高。
85.二聚体的量优选为7摩尔%或更低,更优选5摩尔%或更低。
86.一个实施方案涉及通过如上文进一步概述的方法制备的包含10至20个碳原子,优选14至16个碳原子和最优选14个碳原子的α-烯烃及其混合物的异构化产物。
87.一个实施方案涉及通过包括以下步骤的方法制备的包含10至20个碳原子,优选14至16个碳原子和最优选14个碳原子的α-烯烃及其混合物的异构化产物:
88.(a)在反应区中,优选在固定床反应器中,提供催化剂;
89.(b)将所述反应器惰性化并调整反应器的温度到希望的反应温度;
90.(c)通过控制温度、压力和重时空速(whsv),使所述α-烯烃首先经过预热区,和然后经过所述反应区,优选从底部到顶部而连续进料;
91.(d)任选地,在使用淤浆反应器的情况下,在反应出口从所述反应混合物中移除所述催化剂;
92.(e)在所述反应区后将所述反应混合物冷却到希望的温度以进行进一步加工;
93.(f)任选地,收集经异构化的产物;和/或
94.(g)任选地,去除不希望的副产物,例如通过蒸馏、精馏、真空蒸馏或膜分离。
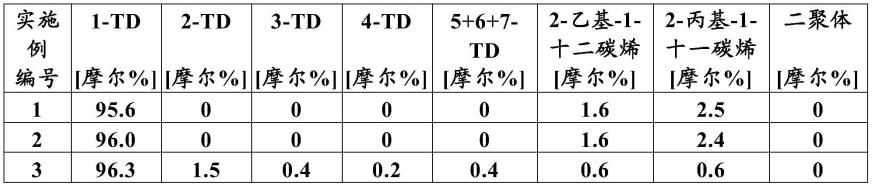
附图说明
95.图1:在所有反应器和实验中的1-十四碳烯转化率的比较。分析数据是通过使用
13
c-nmr方法测定的。
具体实施方式
96.下文中将通过实施例和对比例详细举例说明本发明,但无意将本发明的概念限制于这些特定的实施方案。
97.实验部分
98.十四碳烯异构体分布的
13
c-nmr波谱测定
99.使用离线
13
c-nmr波谱法测定和定量由α-十四碳烯的异构化制备的异构体。所述测定是在添加乙酰丙酮铬作为弛豫助剂的情况下在cdcl3上进行的。使用双键信号进行评价。
100.在所选择的条件下,所得的5-、6-和7-十四碳烯无法区分,因此作为归并在一起的值给出。
101.为了进一步确认信号的多重性,特别是亚甲基和季碳原子的多重性,还记录了
13
c-dept波谱。
102.硅-铝混合氧化物的制备
103.将由45kg/h的ch3sicl3和15kg/h的sicl4组成的混合物的蒸气和0.6kg/h的氯化铝的蒸气借助于作为载气的氮气彼此分开地引入到混合室中。在燃烧器的混合室中将所述蒸气与14.6标准立方米/小时的氢气和129标准立方米/小时的经干燥空气混合,经由中心管进料,在所述中心管末端将反应混合物点燃,进入到水冷却的火焰管中并在那里燃烧。随后将形成的粉末沉积在过滤器中,并用处于400至700℃的水蒸气处理。所述粉末含有99重量%的二氧化硅和1重量%的氧化铝。bet表面积为173m2/g。dbp值为326g/100g混合氧化物。
104.为了测定在具有约5nm厚度的表面层中的初级粒子的重量比(al2o3/sio2)
表面
,采用xps分析。这导致重量比(al2o3/sio2)
表面
为0.0042。在总体初级粒子中的重量比(al2o3/sio2)
总体
的测定是通过对粉末实施的x-射线荧光分析进行的。其显示出重量比(al2o3/sio2)
总体
为0.010。这导致(al2o3/sio2)
总体
/(al2o3/sio2)
表面
的值为2.4。
105.以间歇模式实施的异构化反应
106.对于每个实验,在配备有内部搅拌器的电加热钢制高压釜中填充20g 1-十四碳烯(进料)和1g催化剂。在关闭反应器后,通过将氮气进料而交换在所述反应器中的气相,并将压力调节到约5巴。随后,开始加热所述反应器,并搅拌反应混合物直到希望的反应结束。
107.在反应停止后,通过
13
c-nmr波谱法测量对以间歇模式实施的实验进行详细分析。
108.以连续固定床操作模式实施的异构化反应
109.以连续操作模式实施的实验在两个装置中同时进行,每个装置都由进料容器、hplc泵、预热区、位于加热炉中的两个相继的管式固定床反应器和产物容器组成。每个装置都装填有6g新鲜催化剂。进料由α-十四碳烯组成,并将其首先经由所述hplc泵泵送穿过所述预热区,在所述预热区将进料液体加热到反应温度,和然后从底部到顶部穿过第一个管式固定床反应器,和随后穿过第二个管式固定床反应器。这确保了所述反应器被液体完全充满。在所述反应区之后将反应混合物冷却到环境温度,并将其贮存在产物罐中。所述进料罐以及所述产物罐都用氮气吹扫。
110.反应条件:
111.whsv值=3.1至4.7h-1
112.最大催化剂加载量=12g
113.进料流速=0.8至1.2ml/分钟
114.压力=大气压
115.通过
13
c-nmr定期分析所述连续操作模式的反应产物以测定c
14-烯烃的分布。通过对相同样品进行两次测量来确保所述分析的一致性。
116.1-十四碳烯纯度
117.在开始所述实验之前,对用作进料的1-十四碳烯进行分析以测定初始浓度分布。结果在表1中示出。术语“十四碳烯”在整个表格中被缩写为“td”。
118.表1:所用的1-十四碳烯的初始组成。
[0119][0120]
1-十四碳烯的初始浓度为约96摩尔%,并且主要副组分为2-乙基-1-十二碳烯和2-丙基-1-十一碳烯。支化烃的总浓度为约4摩尔%,并且没有检测到二聚体。
[0121]
催化材料
[0122]
将根据本发明的硅-铝混合氧化物材料(催化剂3)的性能与已知在异构化反应中具有活性的催化参比材料(催化剂1和催化剂2)进行比较。
[0123]
催化剂1(参比):sapo-39
[0124]
催化剂2(参比):amberlyst 15
[0125]
催化剂3(本发明):硅-铝混合氧化物
[0126]
以间歇模式操作实施的性能测试
[0127]
表2中总结了以间歇模式实施的所有实验的实验条件。采用参比催化剂的实施例用符号“*)”标记。术语“十四碳烯”在整个表格中被缩写为“td”。
[0128]
表2.以间歇模式实施的实验的反应条件。
[0129][0130]
表2概述了以间歇模式运行的不同反应所使用的反应条件。首先,进行盲测以确定反应器壁的影响和使用的α-c14烯烃的热稳定性(参见实施例4)。
[0131]
使用不同的反应条件来测定所选催化剂的催化性能,从而确定用于异构化反应的最佳催化剂,找到对于该催化剂的最佳反应条件,并确定不同进料品质对催化性能的影响。进料的量为约20g,和催化剂的量在0.99至1.01g之间(参见实施例5-10)。比较了在300分钟和570分钟下的两种不同反应时间。
[0132]
对于本发明的催化剂,反应温度在130至245℃的范围内变化(参见实施例11-14)。在每个实施例中,将约1g催化剂和20g 1-十四碳烯填充到所述反应器中。
[0133]
对于本发明的催化剂,反应时间进一步在56分钟至335分钟的范围内变化(参见实施例15-20和12)。在每个实施例中,将约1g催化剂和20g 1-十四碳烯填充到所述反应器中。
[0134]
间歇操作实施例的结果
[0135]
下表3显示了上述实施例的转化率和产物分布的结果。
[0136]
表3:在间歇反应中的c
14-烯烃的双键分布的结果。
[0137][0138]
盲测
[0139]
盲测的结果表明,α-c14烯烃在最高至200℃时是热稳定的,因为1-十四碳烯的摩尔分数仅略微降低至92.2摩尔%(参见实施例4)。支化烃的浓度保持在约4摩尔%不变,并且没有观察到二聚体的形成。
[0140]
催化剂变化
[0141]
实施例5-7比较了通过使用不同催化剂实现的转化率和由此获得的双键分布。催化剂2(实施例5)的反应温度为110℃,并且催化剂3(实施例6)和催化剂1(实施例7)的反应温度为200℃,并且反应时间分别为300分钟。对于催化剂2的使用,必须选择较低的温度,因为amberlyst树脂在较高温度下倾向于分解,并且在这种条件下观察到不利的二聚体形成。另一方面,另外两种催化剂在110℃下的转化相当缓慢。
[0142]
实施例5、6和7的1-十四碳烯转化率分别为87.6%、98.7%和9.5%。用催化剂2进行的异构化显示出2-十四碳烯的烯烃浓度最高(49.6摩尔%)。随着碳链中双键位置的碳数增加,相应内烯烃的浓度降低。发现5+6+7-十四碳烯的浓度最低。
[0143]
对于催化剂3,在这些条件下发现了最高的内烯烃异构体浓度,其中分布接近平衡。在平衡条件下,人们会预期每种异构体的含量为14.7摩尔%。实验上,2-、3-和4-异构体几乎达到了这个值。对于5+6+7-十四碳烯的归并在一起的异构体,获得了41.9摩尔%的浓度,其与在平衡时计算的44.1%形成对比。同时,二聚体的浓度达到中等的10.5摩尔%。
[0144]
催化剂1显示出最低的1-十四碳烯转化率。在应用的反应时间之后,1-十四碳烯的残留浓度为86.5摩尔%。2-十四碳烯的摩尔分数的量为8.8摩尔%;所有其它内烯烃的浓度均低于1.0摩尔%。
[0145]
实施例8-10是实施例5-7的重复,通过将反应时间从300分钟增加到570分钟以研究反应时间对平衡组成的影响。由于反应时间较长,当使用催化剂2(实施例10)时,1-十四
碳烯的转化率增加到98.1%。更多内部双键位置异构体的浓度也增加了,但不能达到平衡组成。发现2-十四碳烯的内烯烃浓度最高(34.6摩尔%),并且二聚体的摩尔分数增加了2.3倍。
[0146]
通过使用催化剂3,增加反应时间仅略微改变了内烯烃的异构体组成。然而,二聚体的浓度增加了2倍。使用催化剂1,观察到产物组成没有显著变化。
[0147]
温度变化
[0148]
对于本发明的催化剂,进行了另外的实施例以分别显示1-td转化率和异构体分布的温度依赖性(参见实施例6和实施例11-14)。在130℃下(实施例11),1-十四碳烯的转化率仅为17.6%。发现2-十四碳烯的内烯烃浓度最高(15.5摩尔%)。其它内烯烃的摩尔分数可以忽略不计。在175℃(实施例12)和200℃(实施例6;更短的反应时间)下的结果显示出约99%的几乎完全的转化,并且内烯烃在碳链上的分布几乎相等,即满足平衡条件。随着反应温度的进一步升高,异构体组成不再显著变化(即除α位外,十四碳烯中双键位置的分布相等),但不希望的二聚体形成显著增加。在245℃时,大部分十四碳烯转化为二聚体。
[0149]
反应时间变化
[0150]
当改变反应时间时(参见实施例12和15-20),1-十四碳烯的转化率随着反应时间的增加而增加。56分钟后,1-十四碳烯的转化率为56.1%(实施例15)并在335分钟后增加到99%(实施例12)。然而,随着反应时间的增加,二聚体的摩尔分数也从4.8摩尔%增加到23.8摩尔%。在所有实验中,都发现2-十四碳烯内烯烃的摩尔分数最大,表明双键位置的化学平衡尚未达到。只有在335分钟时(实施例12),内烯烃的浓度几乎相等地分布在碳链长度上。然而,在这种情况下,二聚体的浓度已经非常高了(23.8摩尔%)。
[0151]
以连续模式操作的性能测试
[0152]
在连续固定床操作模式下,考察本发明催化剂的长期稳定性。使用不同的参数组进行四种不同的测试。两个反应器同时运行,并选择两种不同的流速作为初始参数。
[0153]
运行1a(实施例21)
[0154]
在运行1a中,1-十四碳烯以1ml/分钟流过填充有本发明催化剂的反应器。重时空速的量为3.88h-1
。将反应器加热至180℃。11天后,将温度降至170℃,并且其它参数保持恒定。18天后,将流速降至0.8ml/分钟(导致whsv为3.1h-1
)。其它参数保持恒定。总运转时间为32天。表4显示了反应条件和所得1-十四碳烯的转化率。
[0155]
表4:运行1a中的1-十四碳烯的反应参数和转化率x。
[0156][0157]
下表5中报告了运行1a中取样的不同异构体的产物分布。
[0158]
表5:运行1a中的c
14-烯烃的双键分布结果。
[0159][0160]
在运转4小时后,在反应器出口处获得了96.7%的高转化率水平。
[0161]
异构体由高水平的内部双键位置组成,2-位略微占主导地位,占25.3%。随着运转时间的增加,在运行1a中观察到催化剂的轻微失活,这可以从降低的转化率和各个内部双键异构体的较小平衡浓度中看出。当将反应温度降低到170℃时,转化率进一步缩减,2-位的双键位置变得更加占主导地位。随着运转时间增加,失活也会增加。18天后,将进料速率降低到0.8ml/分钟(whsv=3.1h-1
),导致转化率略微增加到89.9%,这是由于在催化剂床中更长的停留时间导致的。然而,异构体组成没有受到显著影响。在所有条件下,二聚体形成率是低的,并且位于第4天的6.6%至32天后的4.3%之间。
[0162]
运行1b(实施例22)
[0163]
在运行1b中,1-十四碳烯以0.9ml/分钟流过填充有本发明催化剂的反应器。重时空速的量为3.49h-1
。将反应器加热到180℃。6天后,将流速增加到1.2ml/分钟(whsv=4.65h-1
)。其它参数保持恒定。11天后,将反应器温度降至170℃。其它参数保持恒定。总运转时间为32天。下表6显示了反应条件和所得1-十四碳烯的转化率。
[0164]
表6:运行1b中的1-十四碳烯的反应参数和转化率x。
[0165][0166]
下表7中报告了运行1b中取样的不同异构体的产物分布。
[0167]
表7:运行1b中的c
14-烯烃的双键分布结果。
[0168][0169]
在运转4小时后,在反应器出口处获得了97%的高转化率水平。
[0170]
异构体由高水平的内部双键位置组成,接近于平衡组成,2-位略微占主导地位,占21.6%。随着运转时间的增加,在运行1b中也观察到催化剂的轻微失活,这可以从降低的转化率和各个内部双键异构体的较小平衡浓度中看出。在第6天将进料流增加到1.2ml/分钟后,转化率按预期下降,并且异构体组成向2-位移动。当在11天后将反应温度降低到170℃时,转化率进一步缩减,2-位的双键位置变得更加占主导地位。18天后,将进料速率降低到0.8ml/分钟(whsv=3.1h-1
),导致转化率略微增加到91.6%,这是由于在催化剂床中更长的停留时间导致的。然而,异构体组成没有受到显著影响。在所有条件下,二聚体形成率是低的,并且位于第4天的7.5%至32天后的4.7%之间。
[0171]
运行2a(实施例23)
[0172]
在运行2a中,1-十四碳烯以1ml/分钟流过填充有本发明催化剂的反应器。重时空速的量为3.88h-1
。将反应器加热至180℃。8天后,将温度降至170℃,并且其它参数保持恒定。总运转时间为25天。下表8显示了反应条件和所获得的1-十四碳烯的转化率。
[0173]
表8:运行2a中的1-十四碳烯的反应参数和转化率x。
[0174][0175]
下表9中报告了运行2a中取样的不同异构体的产物分布。
[0176]
表9:运行2a中的c
14-烯烃的双键分布结果。
[0177][0178]
在运转4小时后,在反应器出口处获得了93.8%的高转化率水平。
[0179]
异构体由高水平的内部双键位置组成,2-位略微占主导地位,占27.3%。随着运转时间的增加,观察到催化剂的轻微失活,这可以从降低的转化率和各个内部双键异构体的较小平衡浓度中看出。当将反应温度降低到170℃时,转化率进一步缩减,2-位的双键位置变得更加占主导地位。随着运转时间增加,失活也会增加。25天后,71.5%的1-十四碳烯被转化。在所有条件下,二聚体形成率是低的,并且位于第4天的5.9%至25天后的4%之间。
[0180]
运行2b(实施例24)
[0181]
在运行2b中,1-十四碳烯以0.9ml/分钟流过填充有本发明催化剂的反应器。重时空速的量为3.49h-1
。将反应器加热到180℃。4天后,将流速增加到1.2ml/分钟(whsv=4.65h-1
)。其它参数保持恒定。8天后,将反应器温度降至170℃。其它参数保持恒定。总运转时间为25天。下表10显示了反应条件和所获得的1-十四碳烯的转化率。
[0182]
表10:运行2b中的1-十四碳烯的反应参数和转化率x。
[0183][0184]
下表11中报告了运行2b中取样的不同异构体的产物分布。
[0185]
表11:运行2b中的c
14-烯烃的双键分布结果。
[0186][0187]
在运转4小时后,在反应器出口处获得了96.1%的高转化率水平。
[0188]
异构体由高水平的内部双键位置组成,接近于平衡组成,2-位略微占主导地位,占19.7%。在第4天将进料流增加到1.2ml/分钟后,转化率没有显著降低,但异构体组成向2-位移动(23.6%)。当在8天后将反应温度降低到170℃时,转化率进一步缩减,2-位的双键位置变得更加占主导地位。在所有条件下,二聚体形成率是低的,并且位于第4天的8.5%至25天后的3.6%之间。
[0189]
总结:
[0190]
根据本发明,在使用硅-铝混合氧化物催化剂时,反应温度和平均whsv是异构化方法中的重要参数。四次运行所获得的结果表明,约180℃的反应温度导致从所用的α-烯烃转化成内烯烃的转化率约为95%,其中内烯烃在碳链上的分布是良好的并且二聚体分数为约6.5至8.5摩尔%。
[0191]
通过将反应温度降低到170℃,二聚体分数可以被降低到远低于5摩尔%。
[0192]
随着时间的推移催化活性的降低,内烯烃的平衡组成向较低的双键位置移动。所用的催化剂在32天的长期稳定性测试后表现出良好的性能。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1