一种α-羰基磺酸酯的合成方法及其使用的催化剂
一种
α
‑
羰基磺酸酯的合成方法及其使用的催化剂
技术领域
1.本发明属于精细化学品绿色合成,具体涉及一种利用氮和碳共掺杂nb2o5催化剂、其制备方法以及使用所述催化剂在可见光驱动下合成α
‑
羰基磺酸酯中的方法。该方法采用绿色的氧气作为氧化剂,使用可见光驱动在温和条件下实现了α
‑
羰基磺酸酯的高效合成。
背景技术:
2.α
‑
羰基磺酸酯是一类重要有机合成中间体,广泛应用于重要医药活性中间体的制备。相对其广泛的应用,制备方法的开发相对滞后。目前为止,基本采用过氧化物、高价碘化合物或过硫化物作为氧化剂使用炔烃、酮等底物在苛刻条件下来完成的。而且存在活性低、选择性差、底物适用范围局限等弊端。因此,开发一种条件温和、底物适用性广泛的催化剂和催化体系具有重要的研究意义实用价值。
3.磺酸酯(ots)作为一种重要的合成砌块在有机合成中被广发的应用。α
‑
羰基磺酸酯骨架的转化和修饰也是得益于ots良好的反应活性和适用性。
4.凭借α
‑
羰基磺酸酯良好的反应活性和重要的应用价值,其广泛应用于含氮、氧、硫等杂子的杂环制备反应中。
5.尽管α
‑
羰基磺酸酯在有机合成领域具有重要的应用价值并且近几年应用方法和策略也被进一步开发。但是,相对应用方面的探索,对其制备方法的开发相对滞后。目前的制备方法往往是使用过氧化物、高价碘化合物、过硫化物等强氧化剂以炔烃或者酮作为底物在相对剧烈的条件下进行的。使用氧气作为氧化剂无论是对于基础研究还是工业应用都是最为理想的氧化策略。基于α
‑
羰基磺酸酯的重要应用价值和目前制备方法存在的弊端,我们开发出具有lewis性和氧化性的双功能nb2o5催化剂并将其应用于烯烃需氧氧化制备α
‑
羰基磺酸酯反应当中,对于该重要合成砌块的制备和双功能性催化剂的开发都具有重要的研究价值。
技术实现要素:
6.针对上述现有技术中的问题,本发明的目的在于开发一种用于合成α
‑
羰基磺酸酯的催化剂,其制备过程简单、原料廉价易得,并且催化过程绿色,环境友好。
7.本发明的另一目的在于提供一种制备根据本发明所述的催化剂的方法。
8.本发明的又一目的在于提供使用根据本发明所述催化剂制备α
‑
羰基磺酸酯的方法。
9.本发明的再一目的在于提供根据本发明所述的催化剂在制备α
‑
羰基磺酸酯的用途。
10.根据本发明的第一方面,提供了一种用于合成α
‑
羰基磺酸酯的氮和碳共掺杂的nb2o5催化剂,其中,基于催化剂的总重量,所述氮的含量0.01wt%~1wt%,优选0.02wt%~0.8wt%,更优选0.03wt%~0.65wt%;所述碳的含量为0.01wt%~1.5wt%,优选为0.01wt%~1wt%,更优选为0.015wt%~0.85wt%。
11.根据本发明的第二方面,提供了一种制备所述催化剂的方法,其包括如下步骤:
12.1)使含碳和氮的前驱体与含铌的前驱体在水和醇的混合溶剂中混合,其中氮与铌的摩尔为1:1~6,优选为1:2~4,最优选1:3;氮与碳的摩尔比为1:0.25~4,优选1:0.3~3;其中混合溶剂与含铌的前驱体的重量比为10~50:1,优选15~40:1
13.2)使步骤1)中得到的产物置于密封水热反应釜中进行水热过程;
14.3)将步骤2)得到产物通过分离、洗涤、干燥后在200℃~600℃,更优选250℃~550℃的温度下进行煅烧。
15.在步骤1)中,
16.优选地,所述含碳和氮的前驱体为分别含碳的前驱体和含氮的前驱体的混合物或者同时含碳和氮的前驱体;更优选为选自2
‑
甲咪唑、尿素、三聚氰胺和乙醇胺中的一种或多种。
17.优选地,所述含铌的前驱体为铌盐,更优选选自草酸铌和氯化铌中的一种或多种。
18.优选地,在步骤1)中的混合溶剂中的水与醇的体积比为1:0.1~10;更优选为1:1~6。
19.优选地,所述醇为具有1至6个碳原子的脂肪醇,更优选为乙醇或丙醇。
20.优选地,在20℃~70℃,更优选在40℃~70℃的温度下进行步骤1)中的混合。
21.在步骤2)中,
22.优选地,水热过程的温度为120℃~250℃,更优选160℃~200℃,最优选为180℃。
23.优选地,水热过程的时间为12小时~36小时,优选18~28小时。
24.在步骤3)中,
25.优选地,煅烧时间为1~5小时,优选1.5~3小时。
26.优选地,升温速率为2℃/min~10℃/min,优选5℃/min。
27.优选地,所述分离通过离心来进行,优选地,离心转速为3000~7000rpm,离心时间5~15分钟,优选5000rpm,10分钟离心时间。
28.优选地,使用乙醇反复洗涤3次,于60℃下真空干燥12小时。
29.根据本发明的第三方面,提供了一种制备α
‑
羰基磺酸酯的方法,其包括如下步骤:
30.在氧气氛围下,使烯烃与亚磺酸盐分散于溶剂中,在根据本发明的氮和碳共掺杂的nb2o5催化剂的存在下,在光照条件下反应得到α
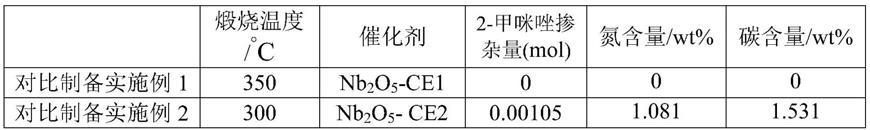
‑
羰基磺酸酯。
31.优选地,所述烯烃为芳香族的末端烯烃、脂肪族的末端烯烃或内烯烃,更优选为未取代的或在苯环上的氢被取代的苯乙烯。
32.优选地,所述亚磺酸盐为取代或未取代的芳基亚磺酸盐,更优选为取代或未取代的苯基亚磺酸盐;进一步更优选地为取代或未取代的苯亚磺酸碱金属盐或取代或未取代的苯亚磺酸碱土金属盐。
33.优选地,所述烯烃与芳基亚磺酸盐的摩尔比为1:2~6。
34.优选地,所述催化剂的质量与所述烯烃的质量比为0.5~3:1。
35.优选地,所述取代的芳基亚磺酸盐中的取代基为选自c
1~10
烷基、卤素、酯基、氰基和三氟甲基中的一种或多种。
36.优选地,在苯环上的氢被取代的苯乙烯的取代基为选自c
1~10
烷基、卤素、酯基、氰基和三氟甲基中的一种或多种。
37.优选地,所述溶剂为选自乙腈、四氢呋喃、二氯甲烷和三氟甲苯中的一种或多种,优选为三氟甲苯。
38.优选地,所述烯烃的浓度为0.01~1mol/l,更优选0.01~0.5mol/l。
39.优选地,所述光照条件为使用的光源为5~30w,更优选为5~10w的led蓝光灯。
40.优选地,反应时间12~48小时,更优选为18~36小时。
41.根据本发明的第四方面,提供了根据本发明所述的催化剂在制备α
‑
羰基磺酸酯的用途。
42.本发明所具有的优点:
43.1、催化剂制备过程简单、原料廉价易得;
44.2、催化过程绿色,环境友好;
45.3、氧气为唯一氧化剂;
46.4、底物适用范围广泛;
47.5、所得产物在医药和材料中间体制备中具有重要应用。
附图说明
48.图1为根据本发明的制备实施例1至4以及对比制备例1得到的催化剂紫外
‑
可见光漫反射测试结果。
49.图2为根据本发明的制备实施例1至4以及对比制备例1得到的催化剂稳态荧光寿命测试结果。
50.图3为根据本发明的制备实施例1至4以及对比制备例1得到的催化剂xrd测试结果。
51.图4为根据本发明的制备实施例2得到的催化剂的核磁氢谱谱图。
52.图5为根据本发明的制备实施例2得到的催化剂的核磁碳谱谱图。
具体实施方式
53.下面结合具体实施例对本发明进行详细说明。在进行描述之前,应当理解的是,在本说明书和所附的权利要求书中使用的术语不应解释为限制于一般含义和字典含义,而应当在允许发明人适当定义术语以进行最佳解释的原则的基础上,根据与本发明的技术方面相应的含义和概念进行解释。因此,这里提出的描述仅仅是出于举例说明目的的优选实例,并非意图限制本发明的范围,从而应当理解的是,在不偏离本发明的精神和范围的情况下,可以由其获得其他等价方式或改进方式。
54.本发明以氮和碳共掺杂nb2o5半导体材料作为催化剂,用于烯烃需氧氧化制备α
‑
羰基磺酸酯,反应条件温和,氧气作为氧化剂,不需要使用其它氧化剂。所述催化剂的所有原料资源丰富,价格低廉,且催化剂可循环使用不失活,对空气、水和热都很稳定。根据本发明的光催化剂和催化体系,烯烃需氧氧化制备α
‑
羰基磺酸酯产率达60%以上。
55.表征所用仪器:
56.1)紫外
‑
可见光漫反射光谱仪:型号为perkinelmer lambda 365uv
‑
vis,生产厂家为珀金埃尔默公司。
57.2)稳态荧光光谱仪:型号为hitachi f
‑
4600spectrofluorometer,生产厂家为
hitachi日立公司。
58.3)元素分析仪:型号为vario
‑
el
‑
cube,生产厂家为elementary公司
59.4)xrd粉末衍射仪:型号为bruker d8 advance diffractomete,生产厂家为bruker公司。
60.5)核磁共振波谱仪:型号为drx
‑
400生产厂家为bruker公司。
61.制备实施例1
62.氮和碳共掺杂nb2o5催化剂的制备。
63.使用0.0345g的2
‑
甲咪唑(0.00042mol)作为碳源和氮源,使用1.02g草酸铌(0.0019mol)作为原料,使用水和乙醇体积混合比例为0.3/1(总体积20ml)的混合溶液在60℃下进行物理混合。水热过程温度为180℃,水热时间为24小时。离心、洗涤、干燥后进行煅烧。从室温开始升温,升温速率为5℃/min,高温煅烧温度为250℃,并在该温度下持续煅烧2小时。待管式炉降到室温后将样品拿出,即得到氮和碳共掺杂nb2o5催化剂。所得催化剂通过元素分析测定含氮量为0.624wt%,含碳量为0.793wt%。
64.制备实施例2至4
65.除了煅烧温度如下表1中所示之外,与制备实施例1相同的步骤制备催化剂,并且结果示于表1中。
66.对比制备实施例1至2
67.除了使用作为碳源和氮原的2
‑
甲咪唑的量和烧结温度如表2中所示之外,与制备实施例1相同的步骤制备催化剂,并且结果示于表2中。氮和碳掺杂量示于下表2中。
68.表1
[0069] 煅烧温度/℃催化剂氮含量/wt%碳含量/wt%制备实施例1250nc
‑
nb2o5‑
2500.6240.793制备实施例2350nc
‑
nb2o5‑
3500.1120.582制备实施例3450nc
‑
nb2o54500.0930.404制备实施例4550nc
‑
nb2o5‑
5500.0340.015
[0070]
表2
[0071][0072]
测试
[0073]
将制备实施例1至4和对比制备实施例1中制备得到的催化剂通过紫外
‑
可见光漫反射测试,所得的结果如图1所示。从图1中可以看出,根据制备实施例1至4制备的催化剂在可见光区有明显的吸收。
[0074]
对根据制备实施例1至4和对比制备实施例1中制备的催化剂进行稳态荧光光谱测试,结果如图2所示。根据图2可以看出,根据本发明的制备实施例1至4制备的催化剂中的氮和碳掺杂能有效抑制电子复合,从而提高反应效率。
[0075]
另外,根据本发明的制备实施例2得到的催化剂的xrd图谱、核磁氢谱谱图和核磁碳谱谱图分别示于图4和图5中。
[0076]
对比例1至2
[0077]
在密闭反应条件下加苯乙烯0.2mmol、对甲基苯亚磺酸钠0.6mmol、使用在对比制备例1至2中得到的催化剂20mg、三氟甲苯2ml,在常压氧气氛围中,5w蓝光led光照射下,于室温反应24小时,过滤反应液,硅胶柱层析即得2
‑
氧代
‑2‑
苯基乙基4
‑
甲基苯磺酸酯结构通过nmr核磁测试得以确定。反应结果示于表3中。
[0078]
实施例1至5
[0079]
在密闭反应条件下加苯乙烯0.2mmol、对甲基苯亚磺酸钠0.6mmol、分别使用在制备实施例1至4得到的催化剂20mg、三氟甲苯2ml,在常压氧气氛围中,5w蓝光led光照射下,于室温反应24小时,过滤反应液,硅胶柱层析即得2
‑
氧代
‑2‑
苯基乙基4
‑
甲基苯磺酸酯,结构通过nmr核磁测试得以确定。具体反应情况如下:
[0080][0081]
表3
[0082] 催化剂氮含量/%碳含量/%产率/%对比例1nb2o5‑
ce10011对比例2nb2o5‑
ce21.0811.53113实施例1nc
‑
nb2o5‑
2500.6240.79377实施例2nc
‑
nb2o5‑
3500.1120.58283实施例3nc
‑
nb2o5‑
4500.0930.40463实施例4nc
‑
nb2o5‑
5500.0340.01560
[0083]
实施例5至12
[0084]
与实施例1操作和步骤相同,改变烯烃和亚磺酸钠(即底物)的种类,得到对应的α
‑
羰基磺酸酯类化合物(产物)、转化率均>99%、产率60~90%不等,具体如表4所示:
[0085]
表4
[0086][0087][0088]
实施例13
[0089]
催化剂循环
[0090]
以苯乙烯和对甲基苯亚磺酸钠反应制备2
‑
氧代
‑2‑
苯基乙基4
‑
甲基苯磺酸酯作为模板反应进行催化剂循环实验,其步骤是:
[0091]
在密闭反应条件下加苯乙烯0.2mmol、对甲基苯亚磺酸钠0.6mmol、在制备实施例2中得到的催化剂20mg、三氟甲苯2ml,在氧气氛围中,5w蓝光led光照射下,于室温反应24小时,过滤反应液,对反应液进行气相色谱分析。将反应液离心(10000rpm,15min),将上清液移除,随后加入5ml乙醇、离心移除上清液,以上操作重复3次,所得固体在真空干燥箱内40℃下干燥12h,以备下一轮催化剂循环使用。在循环实验证实根据本发明的催化剂在重复使用4次后仍然能保持>99%的转化率和>99%的产物选择性。
[0092]
以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。除非特别说明,以下实施例中使用的试剂和仪器均为市售可得产品。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1