一种舍曲林侧链氨基结构衍生物及其制备方法和应用
1.本发明属于医药技术领域,特别涉及一类舍曲林侧链氨基结构的衍生物及其制备方法和应用。
背景技术:
2.舍曲林是一种选择性的血清素再摄取抑制剂,除了在抑郁症中的应用外,近年来有关其在肿瘤治疗中的应用价值的报道也显着增加。例如,据报道舍曲林可以诱导人前列腺癌细胞pc3中的er钙释放。此外,舍曲林与翻译控制的肿瘤蛋白相互作用并降低其细胞水平,导致黑色素瘤细胞的迁移特性和集落形成能力下降。2018年xiufeng pang课题组在“舍曲林的重新利用通过诱导自噬作用使非小细胞肺癌细胞对厄洛替尼敏感”一文中确定了舍曲林在治疗非小细胞肺癌(nsclc)中的作用,证明了舍曲林抑制nsclc细胞的活力,显示出厄洛替尼的协同作用。他们对其机理进行了进一步研究,证明了舍曲林通过靶向ampk/mtor信号通路增强了厄洛替尼在nsclc细胞中的治疗效果。velthedrinberg课题组证明舍曲林与纳米药物联合可以调节癌症的多药耐药性。
3.根据2019年美国癌症学会发布的全球癌症统计数据,胃癌在全球范围内都具有较高的发病率(新发癌症中占5.7%,第五位)和死亡率(癌症引起的死亡人数中占8.2%,第三位)。胃癌的治疗方式通常包括手术、化疗或者靶向药物治疗。但是,由于应答率低、且非常容易发生耐药等原因,常规的治疗手段很难达到令人满意的结果。因此,开发更为有效的新型抗胃癌药物,对提高胃癌的治愈率、延长患者的生存期具有重要的意义。而新型抗胃癌药物的开发,则主要需要解决“肿瘤耐药性”这一关键问题。
技术实现要素:
4.本发明的目的是通过对舍曲林侧链进行修饰制备得到的舍曲林衍生物,并且该舍曲林衍生物在用于耐药胃癌细胞时具有增敏效果,以解决当前胃癌治疗中肿瘤细胞存在对于化疗药物的“耐药性”的问题。
5.本发明首先提供了一种舍曲林侧链氨基结构衍生物,结构式如式ⅰ或式ⅱ所示,
[0006][0007]
其中,r1为h或环丙烷基,r2选自烷基、
萘基、烷基胺、环丙烷基、苯基,或者,3号位和/或4号位被烷基、甲氧烷基、卤代烷基、卤基取代的苯基;r3为氢基或1
‑
卤代烷基。
[0008]
在根据本发明的一个实施方案中,当舍曲林侧链氨基结构衍生物的结构式为式ⅰ时,r2为直链烷基或支链烷基。
[0009]
在根据本发明的一个实施方案中,当舍曲林衍生物的结构式为式ⅰ时,r2选自
‑
(ch2)nch3、n=1、2或4,
‑
ch(ch3)m、m=2、3、4、5、6
…
等大于或等于2的整数,或者
‑
(ch2)xc6h5、x=0、1、2、3
…
等整数。
[0010]
本发明同时还提供了上述舍曲林侧链氨基结构衍生物的制备方法,包括:
[0011]
1)将第一中间体化合物溶解于有机溶剂中,惰性氛围下加入修饰基团化合物、钛酸四异丙酯70
‑
100℃条件下反应;
[0012]
2)步骤1)充分反应后加入氰基硼氢化钠于70
‑
100℃下反应;
[0013]
3)在步骤2)反应完全后,加入淬灭剂淬灭;
[0014]
4)经萃取、除水、层析纯化得到第一产物,所述第一产物是结构式为式ⅰ所示的舍曲林衍生物;
[0015]
其中,第一中间体化合物与修饰基团化合物、钛酸四异丙酯、氰基硼氢化钠的当量比为1:2~1:3.375~1:2.5;
[0016]
所述第一中间体结构式如式ⅲ所示,
[0017][0018]
所述修饰基团结构通式如式v所示,
[0019][0020]
其中,r1为h或环丙烷基,r2选自烷基、选自烷基、萘基、烷基胺、环丙烷基、苯基,或者,3号位和/或4号位被烷基、甲氧烷基、卤代烷基、卤基取代的苯基。
[0021]
在根据本发明的一个实施方案中,还包括:制备结构通式为式ⅱ的舍曲林侧链氨基衍生物,具体步骤如下:
[0022]
5)称取适量第二中间体化合物,于惰性氛围保护的容器中将其与适量二氯甲烷混合,冰浴至0℃后加入三乙胺并搅拌混匀,然后滴加结构式为式
ⅵ
的化合物,于室温搅拌至反应完全后以淬灭剂淬灭,以萃取剂萃取后,经柱层析纯化即得式ⅱ化合物;其中,所述第
二中间体化合物的结构式为式ⅳ,所述r3为氢基或1
‑
卤代烷基;
[0023][0024]
优选地,所述第二中间体化合物、三乙胺、式
ⅵ
化合物的摩尔比为1:2:1.5。
[0025]
在根据本发明的一个实施方案中,步骤1)中所述有机溶剂为异丙醇或甲醇。
[0026]
在根据本发明的一个实施方案中,所述淬灭剂为饱和碳酸氢钠溶液、饱和碳酸氢钠或饱和氯化铵。
[0027]
在根据本发明的一个实施方案中,所述萃取剂为二氯甲烷。
[0028]
在根据本发明的一个实施方案中,所述柱层析叶洗脱液为体积比为10
‑
20:1的石油醚与乙酸乙酯的混合液,或者,体积比为1:100的二氯甲烷与三乙胺的混合液。
[0029]
在根据本发明的一个实施方案中,步骤1)中所述第一中间体化合物是通过包括下述步骤的方法制备得到的:
[0030]
将化合物1与三氯化铝混合,在惰性氛围下加入化合物2,并升温至100℃反应,反应完全后冷却至室温,然后以1n盐酸和冰的混合物进行淬灭,然后萃取、除水、旋干后进行柱层析分离,即得第一中间体化合物;
[0031]
其中,化合物1结构式为化合物2结构式为
[0032]
在根据本发明的一个实施方案中,所述第二中间体是通过包括下述步骤的方法实现的:
[0033]
将适量第一中间体化合物和乙酸铵混合,于惰氛围中加入适量的异丙醇,于70
‑
100℃充分反应后再加入硼氢化钠继续反应,至反应完全,冷却至室温,以淬灭剂淬灭,以萃取剂萃取后,经柱层析纯化得到第二中间体化合物;优选地,所述第一中间体化合物与乙酸铵的摩尔比为1:2;
[0034]
本发明还进一步提供了上述的舍曲林衍生物在制备用于治疗胃癌的药物中的应用。
[0035]
本发明中的合成舍曲林结构的衍生物是独创的未有文献报道;
[0036]
本发明中合成的舍曲林结构的衍生物,其中部分化合物能使耐药胃癌细胞增敏与现有技术相比,本发明具有如下特点:
[0037]
本发明公开了给本发明提供的舍曲林衍生物对于胃癌细胞有明显的致敏效果,能够用于耐药性的胃癌细胞的增敏,或用于提升化疗药物对于肿瘤细胞的抑制作用。
附图说明
[0038]
图1为本发明的舍曲林侧链氨基结构衍生物的合成路线示意图;
[0039]
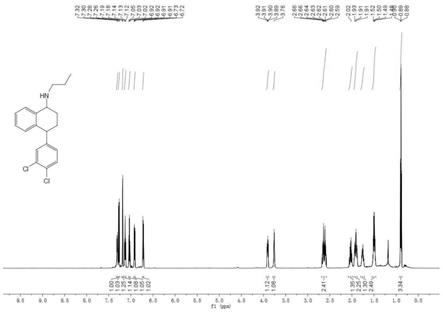
图2为化合物5a的核磁共振氢谱图;
[0040]
图3为化合物6a的核磁共振氢谱图;
[0041]
图4为化合物5k的核磁共振氢谱图;
[0042]
图5为化合物6a的抑制人胃癌细胞耐药株sgc
‑
7901/ddp增殖实验结果图;
[0043]
图6为化合物6b的抑制人胃癌细胞耐药株sgc
‑
7901/ddp增殖实验结果数据图;
[0044]
图7为化合物6c的抑制人胃癌细胞耐药株sgc
‑
7901/ddp增殖实验结果数据图;
[0045]
图8为化合物5t和6d的抑制人胃癌细胞耐药株sgc
‑
7901/ddp增殖实验结果数据图。
具体实施方式
[0046]
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图和实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用以解释本发明,并不用于限定本发明。
[0047]
若未进行特别说明,本实施例中所用的试剂均为分析纯,所有化学反应进度均以薄层色谱进行检测。
[0048]
实施例1:化合物的合成
[0049]
根据本发明的舍曲林侧链氨基结构衍生物的合成流程如图1所示,称取化合物1与三氯化铝于三口烧瓶中,在n2保护的条件下向其中加入化合物2,并升温至100℃反应2.5小时,反应完全后冷却至室温用1n盐酸和冰的混合物进行淬灭,后用二氯甲烷萃取,后进行常规的除水操作,旋干后进行柱层析分离,得中间体化合物;化合物1与三氯化铝、化合物2的当量比为:1:8.157
~
1:1:2.5。
[0050]
1、式ⅰ所示的舍曲林衍生物的合成方法
[0051][0052]
将中间体化合物ⅲ溶解于异丙醇溶液中,n2保护的条件下向其中加入修饰基团化合物、钛酸四异丙酯85℃条件下反应两小时后,向其中加入氰基硼氢化钠在85℃条件下反应,反应至反应完全,反应完全后用饱和碳酸氢钠溶液淬灭,用二路甲烷萃取后进行常规除水操作,旋干后进行柱层析分离,得到结构式如式ⅰ所示的舍曲林衍生物;中间体化合物与修饰基团化合物、钛酸四异丙酯、氰基硼氢化钠的当量比为1:2
~
1:3.375
~
1。
[0053]
所述修饰基团结构式如式v所示。
[0054][0055]
其中,r1为h或环丙烷基,r2选自烷基、选自烷基、萘基、烷基胺、环丙烷基、苯基,或者,3号位和/或4号位被烷基、甲氧烷基、卤代烷基、卤基取代的苯基。
[0056]
2、式ⅱ所示舍曲林衍生物的合成步骤
[0057]
如图1所示的,在获得中间体化合物ⅲ后,还可以进一步合成式ⅱ所示的舍曲林化合物衍生物。
[0058][0059]
称取中间体化合物ⅲ(583.9mg(2mmol))和化合物乙酸铵(311.4mg(4mmol))于三口瓶中抽换气并用n2保护,并向其中加入适量的异丙醇,升温至80摄氏度反应2小时,后再向其中加入化合物硼氢化钠(756.6mg(20mmol))在80℃的条件下反应16小时,冷却至室温,用饱和碳酸氢钠溶液淬灭,并用二氯甲烷萃取,干燥,旋干,并用pe:ea=20:1的洗脱液进行柱层析分离,旋干后得中间体化合物ⅳ。
[0060]
称取中间体化合物ⅳ(200mg(0.68mmol))于烧瓶中抽换气并用氮气保护并加入适量的二氯甲烷。冰浴至0℃后向其中加入三乙胺(137.7mg(1.36mmol))搅拌5分钟,后再向其中滴加结构式为式
ⅵ
的化合物(1.03mmol),并于室温搅拌至反应完全,后用饱和碳酸氢钠溶液淬灭,二氯甲烷萃取,无水硫酸钠干燥,旋干柱层析分离得结构式为式ⅱ的舍曲林侧链氨基衍生物。
[0061]
本发明合成得到的部分舍曲林衍生物列表如表1所示:
[0062]
表1本发明制备得到的舍曲林衍生物展示表
[0063][0064]
[0065]
实施例2编号no.1舍曲林衍生物的合成
[0066]
将称量中间体化合物ⅲ(583.9mg(2mmol))于干燥的放有搅拌子的三口圆底烧瓶中,进行无水无氧处理,后向其中加入正丙胺(0.33ml(4mmol))搅拌5分钟后又向其中加入钛酸四异丙酯(2ml(6.75mmol))于85摄氏度的油浴锅内进行回流2小时,2小时后将其冷却至室温后再向其中加入氰基硼氢化钠(314.2mg(5mmol))并升温至85℃回流至反应完全。反应完全后将反应液冷却至室温后向其中加入适量的饱和的碳酸氢钠溶液进行淬灭,后再用二氯甲烷进行萃取,后用无水硫酸钠进行干燥,抽滤,旋干,得黄色油状物;后进行柱层析分离洗脱剂为石油醚:乙酸乙酯=20:1,收集洗脱液,浓缩至干可得产物为无色油状物收率为47%。
[0067]
利用600mhz核磁氢谱和高分辨确定化合物5a结构式:1h nmr(600mhz,cdcl3),δ7.31(d,j=7.5hz,1h),7.27(d,j=8.2hz,1h),7.19(d,j=2.1hz,1h),7.13(t,j=7.4hz,1h),7.03(t,j=7.5hz,1h),6.92(dd,j=8.2,1.5hz,1h),6.72(d,j=7.7hz,1h),3.91(dd,j=9.0,5.7hz,1h),3.76(s,1h),2.69
–
2.57(m,2h),2.08
–
1.99(m,1h),1.96
–
1.88(m,2h),1.80
–
1.72(m,1h),1.50(dq,j=14.2,7.1hz,2h),0.89(t,j=7.4hz,3h);hrms(esi+):m/z[m+h]+calcd for c
19
h
22
cl2n:334.1124,found 334.1124.
[0068][0069]
实施例3编号no.22舍曲林衍生物的合成
[0070]
将称量中间体化合物ⅲ(583.9mg(2mmol))于干燥的放有搅拌子的三口圆底烧瓶中,进行无水无氧处理,后向其中加入boc保护的乙二胺(0.65ml(4mmol))搅拌5分钟后又向其中加入钛酸四异丙酯(2ml(6.75mmol))于85℃的油浴锅内进行回流2小时,2小时后将其冷却至室温后再向其中加入氰基硼氢化钠(314.2mg(5mmol))并升温至85摄氏度回流至反应完全。反应完全后将反应液冷却至室温后向其中加入适量的饱和的碳酸氢钠溶液进行淬灭,后再用二氯甲烷进行萃取,后用无水硫酸钠进行干燥,抽滤,旋干,得黄色油状物;然后通过柱层析分离,洗脱剂为体积比为10:1的石油醚:乙酸乙酯,收集洗脱液,浓缩至干可得产物为无色油状物,称取无色油状物于放有搅拌子烧瓶中,后向其中加入适量的三氟乙酸与二氯甲烷,并且三氟乙酸:二氯甲烷=1:5,常温搅拌3.5小时,反应完全后用饱和碳酸氢钠溶液淬灭并用二氯甲烷萃取,后用无水硫酸钠干燥,抽滤旋干,后进行柱层析分离,洗脱剂为二氯甲烷:三乙胺=100:1,收集洗脱液,浓缩至干可得产物为无色油状物收率为83%;
[0071]
利用600mhz核磁氢谱和高分辨确定该化合物(代码6a)结构式:1h nmr(600mhz,cdcl3)δ7.39(d,j=7.7hz,1h),7.32(d,j=8.2hz,1h),7.24(s,1h),7.18(t,j=7.4hz,1h),7.08(t,j=7.4hz,1h),6.96(d,j=8.2hz,1h),6.78(d,j=7.7hz,1h),4.01
–
3.94(m,1h),3.81(d,j=4.0hz,1h),2.91
–
2.77(m,4h),2.06(dd,j=21.2,10.1hz,1h),2.02
–
1.91(m,
2
h),1.81(dd,j=17.1,7.4hz,1h);hrms(esi
+
):m/z[m+h]
+
calcd for c
18
h
21
cl2n2:335.1076,found 335.1083.
[0072]
化学反应方程式如下:
[0073][0074]
实施例4编号no.11舍曲林衍生物的合成
[0075]
将称量中间体化合物ⅲ(583.9mg(2mmol))于干燥的放有搅拌子的三口圆底烧瓶中,进行无水无氧处理,后向其中加入对甲氧基苄胺(0.52ml(4mmol))搅拌5分钟后又向其中加入钛酸四异丙酯(2ml(6.75mmol))于85℃的油浴锅内进行回流2小时,2小时后将其冷却至室温后再向其中加入氰基硼氢化钠(314.2mg(5mmol))并升温至85℃回流至反应完全。反应完全后将反应液冷却至室温,然后向其中加入适量的饱和碳酸钠溶液进行淬灭,后再用二氯甲烷进行萃取,后用无水硫酸钠进行干燥,抽滤,旋干,得黄色油状物;后进行柱层析分离洗脱剂为石油醚:乙酸乙酯=20:1,收集洗脱液,浓缩至干可得产物为无色油状物收率为27%;
[0076]
利用600mhz核磁氢谱和高分辨确定化合物(代码5k)结构式:1h nmr(600mhz,cdcl3)δ7.34(dd,j=16.1,6.8hz,4h),7.29(s,1h),7.19(t,j=7.4hz,1h),7.11(t,j=7.4hz,1h),7.01(d,j=8.2hz,1h),6.91(d,j=8.2hz,2h),6.80(d,j=7.7hz,1h),4.00(dd,j=8.6,5.0hz,1h),3.93(d,j=13.0hz,1h),3.87(s,1h),3.81(d,j=12.3hz,4h),2.18(dd,j=20.7,11.2hz,1h),2.06
–
1.99(m,2h),1.85(td,j=11.5,4.2hz,1h);hrms(esi
+
):m/z[m+h]
+
calcd for c
24
h
24
cl2no:412.1229,found412.1234.
[0077]
化学反应方程式如下:
[0078][0079]
实施例5化合物抑制人胃癌细胞耐药株sgc
‑
7901/ddp增殖实验
[0080]
(1)细胞收集
[0081]
a.胃癌细胞耐药株sgc
‑
7901/ddp(购于美国模式培养物集存库(atcc))用完全培养基(含有10%体积胎牛血清和1%体积青链双抗的rpmi
‑
1640培养基)在37℃,5%co2条件
下培养48小时后,显微镜观察细胞生长状态,待细胞融汇度达到80%
‑
90%后从孵箱取出;
[0082]
b.真空泵吸除培养基,用pbs缓冲液洗涤细胞一次,吸出pbs,向培养皿中加入1ml预热至室温的胰酶消化液,37℃孵箱消化2分钟;
[0083]
c.加入5ml预热至37℃的完全培养基终止消化,并用移液枪反复轻柔吹打培养皿底,使细胞脱落形成细胞悬液;
[0084]
d.将细胞悬液转入15ml离心管中,用低速离心机以800rpm转速离心5min后吸除上清,加入10ml完全培养基重悬细胞。
[0085]
(2)细胞铺板
[0086]
a.取96孔板一块,于四周36个孔中各加入pbs缓冲液100μl封板,在中央的60个孔中各加入90μl上述细胞悬液,此时各孔中细胞数为5000个/孔;
[0087]
b.将接种好细胞的96孔板放回37℃孵箱继续培养过夜使细胞贴壁。
[0088]
(3)加药处理
[0089]
a.分别精密称量编号no.22、23、24和25化合物后用二甲基亚砜(dmso)配制成50mmol的药物母液,并用完全培养基(含有10%体积胎牛血清和1%体积青链双抗的rpmi
‑
1640培养基)稀释至10、20、40、80、120、160、200、240μmol/l的系列浓度梯度药液;
[0090]
b.将以上稀释后的药液按照10μl/孔的剂量加入到对应的细胞接种孔中,每个浓度设置3个复孔(此时各孔中化合物终浓度为1、2、4、8、12、16、20、24μmol/l),同时设置阴性对照孔(加入体积分数0.1%的二甲基亚砜完全培养基溶液)和空白对照孔(不含细胞和药液);
[0091]
c.加完药后充分振摇孔板,使药液充分混匀,后放回37℃孵箱继续培养24h。
[0092]
(4)细胞增殖活力检测
[0093]
a.向各孔加入10μl cck
‑
8溶液(cell counting kit
‑
8,购自碧云天,c0039),充分振摇孔板后放回37℃孵箱继续培养2h;
[0094]
b.取出96孔板,用酶标仪在450nm波长处读取各孔od值;
[0095]
c.计算各化合物抑制胃癌细胞耐药株sgc
‑
7901/ddp增殖的半数抑制浓度(ic50)。
[0096]
实验结果如图5
‑
图8所示:所合成的化合物均能发挥抑制胃癌细胞耐药株sgc
‑
7901/ddp增殖的生物学活性。
[0097]
上述发明内容及具体实施方式意在证明本发明所提供技术方案的实际应用,不应解释为对本发明保护范围的限定。本领域技术人员在本发明的精神和原理内,当可作各种修改、等同替换、或改进。本发明的保护范围以所附权利要求书为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1