一种α-烯烃-环烯烃共聚物及其制备方法和应用与流程
一种
α-烯烃-环烯烃共聚物及其制备方法和应用
技术领域
1.本发明提供一种α-烯烃-环烯烃共聚物及其制备方法和应用,更具体而言,提供一种采用非茂金属配合物和助催化剂来制备α-烯烃-环烯烃共聚物的方法以及由此所制备的具有高tg的α-烯烃-环烯烃共聚物及其应用。
背景技术:
2.环烯烃共聚物,简称coc(cyclic-olefin copolymers),是一类由环烯烃加成共聚合而成的高附加值热塑性工程塑料,因其具有高透明性、高玻璃化转变温度(tg)和高耐化学品性等特点而倍受关注。其中,玻璃化温度是受共聚单体比例控制的,环烯烃单元的含量愈高,共聚物的tg愈高。高玻璃化转变温度的coc共聚物具有高产品纯度、透明性和高热变形温度,用于医疗保健领域的应用,在此应用中要求清洁和耐高温消毒操作,及用于光学数据存储如cd和cd-rom,这些应用中低双折射和高模塑重复性是重要的要求。
3.早期环烯烃共聚合采用的是ziegler-natta催化剂,但是ziegler-natta催化剂具有多个活性中心,聚合活性很低,耐极性基团能力差,限制了其应用。随着聚合活性更高茂金属催化剂的出现,对其的研究也日益活跃起来。中国专利申请cn101125901a中公开一种采用茂金属催化剂制备组成分布窄的环烯烃共聚物的方法;中国专利cn102702433b公开了一种采用半茂金属催化剂制备高分子量的乙烯与降冰片烯共聚物;中国专利cn102286126a提供了一种采用茂金属催化剂制备低环烯烃含量的高透明环烯烃共聚物;中国专利cn101613437b公开了一种采用茂金属催化剂制备带极性基团的环烯烃共聚物。与传统的ziegler-natta催化剂相比,茂金属催化剂可根据催化剂和配体的结构来控制共聚物的分子量、立规规整度和共聚单体反应性。
4.近年来,非茂单活性中心催化剂由于具有与茂金属催化剂不同的性能,且易于合成,受到普遍关注。与茂金属化合物相比,非茂金属化合物能提供亲电性更强的活性中心及更开放的配位空间,因此可能具有更高的环烯烃单体插入效率,可以催化高单体比的降冰片稀的共聚合,且共聚物的空间结构也不同,反映在产品的性能上也会有一定的变化。中国专利申请cn1887925a中公开一类非茂金属催化剂,可在低用量助催化剂的作用下催化乙烯与环戊二烯、降冰片烯等环烯烃的共聚合,由该非茂金属催化剂所得的乙烯-环烯烃共聚物和通常的茂金属催化制备的乙烯-环烯烃共聚物相比具有更高的强度和模量,这大大地拓展了共聚物的应用范围,例如对于包装材料在直立性和挺括度有特殊要求的场合。但和通常的茂金属催化乙烯-环烯烃共聚相比,该非茂金属催化剂的聚合活性相对偏低,需要用烷基铝氧烷为助催化剂进行溶液聚合。另一方面,由于昂贵的助催化剂mao(甲基铝氧烷)、mmao(修饰的甲基铝氧烷)或者dmao(干燥的甲基铝氧烷)的大量使用,极大地增加生产成本,同时也大大增加了聚合物中金属的含量,不利于其工业化的应用。
5.因此,在现有技术中,存在以下的技术问题:难以低成本地、工业上有利地以高聚合活性的催化剂来制备表现出优异性质的乙烯-环烯烃共聚物。
技术实现要素:
6.鉴于上述技术问题,本发明人进行了深入地研究,结果发现,通过使用由特定结构的非茂催化剂与烷基铝和硼化合物形成的催化体系,即使不使用现有技术中必须的高价格的铝氧烷类助催化剂的情况下,也可以以高聚合活性制备出α-烯烃-环烯烃共聚物。并且,所制备出的α-烯烃-环烯烃共聚物具有高tg和高透明度的性状,表现出优异的应用前景。
7.不受任何理论限定地,本发明人推测,通过使用有机硼化物和烷基铝作为助催化剂,与非茂金属配合物构成单中心催化体系,能替代烷基铝氧烷,由此,不但不会降低催化剂体系的聚合活性,而且能大大降低生产成本。另外,通过加入适量的链转移剂可以调整聚合产物的分子量,避免聚合中高环烯烃浓度的形成的凝胶产物。
8.通过本发明的方法制备的α-烯烃-环烯烃共聚物表现出高玻璃化转变温度(tg)和高透明度,在产业中的应用更加广泛。并且,本发明的方法中,可以使用烷烃等作为溶液聚合溶剂,聚合条件温和,共聚活性高,这使得本发明方法的制造成本显著降低。从而解决了现有技术中存在的技术问题。
9.具体而言,本发明提供一种α-烯烃-环烯烃共聚物的制备方法,其包括以下步骤:在非茂金属配合物和助催化剂的存在下,使α-烯烃和环烯烃共聚。
10.进而,本发明提供一种α-烯烃-环烯烃共聚物,其是通过本发明的α-烯烃-环烯烃共聚物的制备方法而制备的。
11.进而,本发明提供一种聚合物组合物,其至少包含本发明的α-烯烃-环烯烃共聚物。
12.另外,本发明还提供本发明的α-烯烃-环烯烃共聚物或本发明的聚合物组合物在制造光学部件、包装材料、电子部件、医疗器具中的应用。
13.技术效果通过本发明的α-烯烃-环烯烃共聚物的制备方法,可以在不使用铝氧烷系助催化剂的情况下,低成本且高效地制备α-烯烃-环烯烃共聚物。另外,本发明的制备方法中,可以通过控制烷基铝的比例来方便地调控分子量,由此可以调节共聚物的形态。另外,本发明的α-烯烃-环烯烃共聚物的制备方法可以在惰性有机溶剂中进行,在聚合中不会出现爆聚现象,聚合条件温和,降低了对生产设备的要求。
14.并且,本发明的α-烯烃-环烯烃共聚物中,环烯烃单元含量高,共聚物具有优异的透明性以及高玻璃化转变温度,在产业中的应用范围广泛。
附图说明
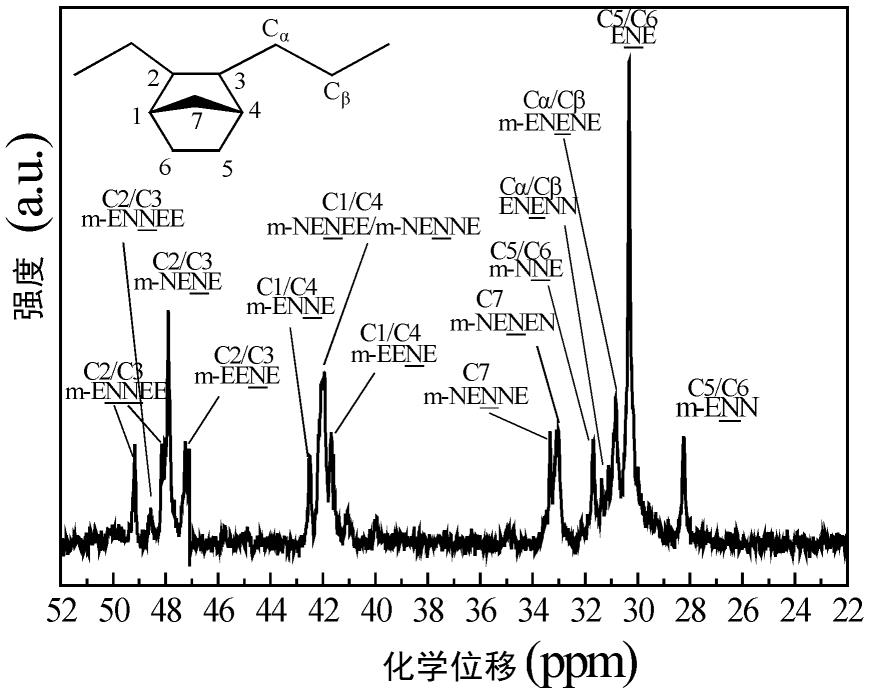
15.图1 为用于示意α-烯烃-环烯烃共聚物中环烯烃含量计算的coc典型核磁共振图谱。
具体实施方式
16.下面对本发明的具体实施方式进行详细说明,但是需要指出的是,本发明的保护范围并不受这些具体实施方式的限制,而是由附录的权利要求书来确定。
17.在本发明的上下文中,除非另有明确定义,或者该含义超出了本领域技术人员的理解范围,3个碳原子以上的烃或烃衍生物基团(比如丙基、丙氧基、丁基、丁烷、丁烯、丁烯
基、己烷等)在未冠以词头“正”时均具有与冠以词头“正”时相同的含义。比如,丙基一般理解为正丙基,而丁基一般理解为正丁基,除非另有明确。
18.另外,本文描述的任何实施方式均可以与本文描述的一种或多种其他实施方式自由结合,由此而形成的技术方案或技术思想均视为本发明原始公开或原始记载的一部分,而不应被视为是本文未曾披露或预期过的新内容,除非本领域技术人员认为该结合是明显不合理的。
19.并且,本发明中,描述同一物性的多组数值范围的端点值可以任意地进行组合,本领域技术人员可以确认这种组合显然是在本发明原始公开或原始记载的一部分,而不应被视为是本文未曾披露或预期过的新内容。
20.在没有明确指明的情况下,本说明书内所提到的所有百分数、份数、比率等都是以重量为基准的,除非以重量为基准时不符合本领域技术人员的常规认识。
21.本发明的α-烯烃-环烯烃共聚物的制备方法包括以下步骤:在非茂金属配合物和助催化剂的存在下,使α-烯烃和环烯烃共聚。由此,制备得到α-烯烃-环烯烃共聚物。
22.本发明中,所述α-烯烃为由下式(a)表示的烯烃。r-ch=ch2ꢀꢀꢀꢀꢀ(a)其中r表示h或者c
1-8
直链或支链烷基,优选h或者c
1-4
直链或支链烷基,更优选h、甲基或乙基。
23.在本发明的一个实施方式中,所述α-烯烃例如可以举出c
2-10
直链或支链烯烃,优选c
2-6
直链或支链烯烃,更优选为c
2-3
链烯烃,进一步优选为乙烯或丙烯。
24.在本发明的一个实施方式中,这些α-烯烃可以单独使用,也可以将两种或多种组合使用。
25.本发明中,所述环烯烃是指单环或多环、且在环上具有双键的烯烃。
26.在本发明的一个实施方式中,环烯烃具体可以举出c
3-20
环烯烃。作为所述c
3-20
环烯烃,具体比如可以举出环丁烯、环戊烯、环戊二烯、环己烯、环己二烯、环庚烯、环庚二烯、环辛四烯、四环十二烯、三环十二烯、三环十一烯、五环十五烯、五环十六烯、8-乙基四环十二烯等单环环烯烃,以及双环戊二烯、降冰片烯、降冰片二烯、、、和等螺环、桥环或稠环式双环或多环环烯烃。作为所述c
3-20
环烯烃,优选环戊烯、环戊二烯、降冰片二烯、双环戊二烯、降冰片烯、乙烯基降冰片烯和乙叉降冰片烯或者四环十二烯。
27.在本发明的一个实施方式中,c
3-20
环烯烃任选进一步被一个或多个(比如1至5个、1至4个、1至3个、1至2个或者1个)c
1-10
直链或支链烃基在合适的位置处取代。作为所述c
1-10
直链或支链烃基,优选c
1-10
直链或支链烷基或者c
2-10
直链或支链烯基,更优选c
1-4
直链或支链烷基或者c
2-4
直链或支链烯基,其中更优选甲基、乙基、乙烯基或者乙叉基。
28.在本发明的一个实施方式中,作为c
3-20
环烯烃,例如可以进一步举出下式(y)所示的化合物。
ꢀꢀꢀꢀꢀ(y)。
29.在式(y)中,基团ra、rb、rc、rd、re、rf、rg、rh可以相同也可以不同,各自独立地选自氢、c
1-10
直链或支链烃基。作为所述c
1-10
直链或支链烃基,优选选自c
1-10
直链或支链烷基或者c
2-10
直链或支链烯基,更优选c
1-4
直链或支链烷基或c
2-4
直链或支链烯基。
30.在本发明的一个实施方式中,在式(y)中,基团ra至基团rh可以相同也可以不同,各自独立地选自氢、c
1-4
直链或支链烷基和c
2-4
直链或支链烯基比如c
2-3
直链或支链烯基,其中优选各自独立地选自氢、甲基、乙基、乙烯基或者乙叉基。
31.在本发明的一个实施方式中,在式(y)中,n为0至6的整数,优选0或1。
32.在本发明的一个实施方式中,在式(y)中,符号代表单键或者双键。
33.在本发明的一个实施方式中,作为所述式(y)所示的化合物,优选降冰片烯、乙叉降冰片烯、乙烯基降冰片烯、降冰片二烯、5-甲基降冰片烯,更优选降冰片烯、乙叉降冰片烯、乙烯基降冰片烯。
34.在本发明的一个实施方式中,所述环烯烃可以单独使用,也可以两种或多种组合使用。
35.本发明中,所述非茂金属配合物为选自下式(i)所示的化合物。ꢀꢀꢀꢀꢀ(i)。
36.在本发明的一个实施方式中,在式(i)中,基团r1、r2、r3、r4可以相同也可以不同,各自独立地选自氢和c
1-6
直链或支链烃基,优选各自独立地选自氢和c
1-6
直链或支链烷基,更优选各自独立地选自氢、甲基、乙基、丙基、丁基、异丁基、仲丁基、叔丁基。在本发明的一个实施方式中,r1、r3表示氢。在本发明的一个实施方式中,r2、r4各自独立地选自氢、甲基、乙基、丙基、丁基、异丁基、仲丁基和叔丁基,优选各自独立地选自氢和叔丁基。
37.在本发明的一个实施方式中,在式(i)中,基团r6、r7、r8、r9可以相同也可以不同,各自独立地选自氢和c
1-6
直链或支链烃基,优选各自独立地选自氢和c
1-6
直链或支链烷基,更优选各自独立地选自氢、甲基、乙基、丙基、丁基、异丁基、仲丁基、叔丁基。在本发明的一
个实施方式中,r7、r9表示氢。在本发明的一个实施方式中,r6、r8各自独立地选自氢、甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基和叔丁基,优选各自独立地选自氢和叔丁基。
38.在本发明的一个实施方式中,在式(i)中,基团r5表示氢或c
1-12
直链或支链烃基,优选氢、c
1-6
直链或支链烷基或c
6-10
芳基,更优选氢、c
1-3
直链或支链烷基或苯基,更优选氢、甲基、乙基、正丙基或异丙基。
39.在本发明的一个实施方式中,在式(i)中,r
10
表示氢或c
1-6
直链或支链烃基,优选氢或c
1-6
直链或支链烷基,更优选氢、甲基和乙基,更优选氢。
40.在本发明的一个实施方式中,符号表示单键或者双键,当表示双键时,n上的h不存在,当表示单键时,n上的h存在。
41.在本发明的一个实施方式中,在式(i)中,基团y是o或s,优选o。
42.在本发明的一个实施方式中,在式(i)中,基团a是s或o,优选s。
43.在本发明的一个实施方式中,在式(i)中,基团m选自元素周期表第iii族到第vi族的金属元素,优选第ivb族金属元素比如钛、锆和铪,更优选钛。
44.在本发明的一个实施方式中,在式(i)中,基团x为卤素,包括氟、氯、溴和碘,其中优选氯或溴。
45.在本发明的一个实施方式中,在式(i)中,符号
‑‑‑‑‑‑
代表配位键。
46.在本发明的一个实施方式中,n为1、2、3、4或5,取决于所述中心金属原子m的价态,优选n为2、3或4。
47.在本发明的一个实施方式中,式(i)所示的非茂金属配合物为选自3-叔丁基亚水杨基2-甲硫基苯胺三氯化钛、水杨基2-甲硫基苯胺三氯化钛、亚水杨基2-苯硫基苯胺三氯化钛、3,5-二叔丁基亚水杨基2-丙硫基苯胺三氯化钛、3-叔丁基亚水杨基2-丙硫基苯胺三氯化钛、3,5-二叔丁基亚水杨基2-巯基苯胺三氯化钛、亚水杨基2-巯基苯胺三氯化钛、亚水杨基2-甲硫基苯胺三氯化钛、亚水杨基2-丙硫基苯胺三氯化钛、3,5-二叔丁基亚水杨基2-甲硫基苯胺三氯化钛、3,5-二叔丁基水杨基2-甲硫基苯胺三氯化钛、3,5-二叔丁基水杨基2-丙硫基苯胺三氯化钛中至少一种。
48.在本发明的一个实施方式中,式(i)所示的非茂金属配合物可以单独使用一种,或者以任意的比例组合使用多种。
49.本发明中,式(i)所示的非茂金属配合物可以通过本领域已知的方法合成得到,也可以通过市售途径得到。
50.在本发明的一个实施方式中,所述式(i)所示的化合物比如可以通过如下的制造方法进行制造。
51.该制造方法比如包括使下式(i-a)所示的化合物与下式(x)所示的化合物发生配位反应,获得所述式(i)所示的化合物的步骤(以下称为配位步骤a)。
ꢀꢀꢀꢀꢀ(i-a)mx4ꢀꢀꢀꢀꢀ(x)。
52.本发明中,式(i-a)中的各基团的定义以及优选的方案与本发明上述式(i)中的定义相同。式(i-a)中,当表示单键时,p表示2,n上的h存在,当表示双键时,p表示1,n上的h不存在。
53.本发明中,在式(x)中,基团m选自元素周期表第iii族到第vi族的金属元素,优选第ivb族金属元素比如钛、锆和铪,其中更优选钛。
54.根据本发明,在式(x)中,基团x为卤素,包括氟、氯、溴和碘,其中优选氯或溴。
55.根据本发明,在进行所述配位步骤a时,所述式(x)所示的化合物与所述式(i-a)所示的化合物的摩尔比一般为0.7-1.5:1,优选0.9-1.3:1,更优选1-1.2:1。
56.根据本发明,所述配位步骤a可以在溶剂的存在下进行。本发明对所述溶剂没有特别的限定,只要其可以溶解所述式(x)所示的化合物与所述式(i-a)所示的化合物但不干扰所述配位反应即可。具体而言,作为所述溶剂,比如可以举出c
5-20
烷烃、c
6-20
芳香烃和c
4-20
脂环烃等,其中优选c
6-12
芳香烃,最优选甲苯、二甲苯、三甲苯。这些溶剂可以单独使用一种,或者以任意的比例组合使用多种。
57.根据本发明,对所述溶剂的用量没有特别的限定,只要是有利于该配位反应进行的任何用量均可以采用,比如可以举出该溶剂和所述式(i-a)所示的化合物的摩尔比为5-200,优选10-100,但并不限于此。
58.根据本发明,所述配位步骤a的反应温度一般为-80-100℃,优选-50-70℃,更优选-30-50℃。
59.根据本发明,所述配位步骤a的反应压力可以是适合该配位反应进行的任何压力,但为了方便实施起见,一般为常压至0.2mpa。
60.根据本发明,所述配位步骤a的反应时间一般为0.1-72小时,优选0.2-48小时,更优选1-24小时。
61.根据本发明,根据需要,为了促进反应的进行,所述配位步骤a的配位反应可以在搅拌(比如搅拌转速为100-1000rpm)下进行。
62.根据本发明,根据需要,所述配位步骤a的配位反应可以在保护性气体的氛围下进行。作为所述保护性气体,比如可以举出非活性气体如氮气等。
63.根据本发明,在所述配位步骤a的配位反应结束后,通过常规的分离方式即可从该
反应获得的混合物中分离出所述式(i)所示的化合物作为反应产物。作为所述的分离方式,比如可以举出过滤或者先过滤再洗涤,以及任选进一步干燥等。或者,根据需要,还可以通过重结晶、柱层析方法或制备色谱等对所述获得的反应产物进行纯化。
64.根据本发明,对于所述过滤、洗涤和干燥的方法并没有特别的限定,可以根据需要使用本领域常规使用的那些。根据需要,所述洗涤一般进行1-6次,优选2-3次。其中,洗涤用溶剂优选与配位反应所使用的溶剂相同,但也可以不同。作为所述干燥,比如可以举出惰性气体干燥法、真空干燥法或者真空下加热干燥法等,其中优选惰性气体干燥法或真空下加热干燥法,最优选真空下加热干燥法。此时,所述干燥的温度范围一般为常温至140℃,干燥时间一般为2-20小时,但并不限于此。
65.本发明中,所述助催化剂为有机硼化合物、和选自烷基铝、烷基铝水解物、卤代烷基铝中的至少一种烷基铝衍生物的混合体系。
66.在本发明的一个实施方式中,所述制备方法中不使用铝氧烷类助催化剂。
67.在本发明的一个实施方式中,所述有机硼化合物为选自烷基硼、芳基硼和硼酸盐中的至少一种。
68.在本发明中,所述烷基硼、芳基硼可以为具有如下通式(b-1)的化合物:b(r)3ꢀꢀꢀꢀꢀ(b-1)其中,三个r基团中的每一个彼此可以相同,也可以不同,各r独立地选自c
1-6
直链或支链烷基和c
6-12
芳基,各基团任选地被一个以上卤素原子、卤代c
1-6
直链或支链烷基或苯氧基取代。所述r优选选自甲基、乙基、丙基、丁基、异丁基、苯基、甲苯基、三氟甲基苯基和五氟苯基。作为烷基硼的具体例,可以列举三甲基硼、三乙基硼、三异丁基硼、三丙基硼或三丁基硼。作为芳基硼的具体例,可以列举三(五氟苯基)硼、三[3,5-双(三氟甲基)苯基]硼。
[0069]
在本发明中,所述硼酸盐可以为具有如下通式(b-2)的化合物:[l]
+ [be4]
m-ꢀꢀꢀꢀꢀ(b-2)其中l为阳离子基团,各个e可以相同或不同,各自独立地选自卤素原子、c
6-12
芳基,所述芳基任选地被一个以上卤素原子、c
1-6
直链或支链烷基、卤代c
1-6
直链或支链烷基、c
1-6
直链或支链烷氧基或苯氧基所取代。所述e优选选自氟、苯基、三氟甲基苯基和五氟苯基。m表示l部分的基团的化合价的数值。
[0070]
[l]
+
部分可以是硼酸盐中常见的阳离子,例如,可以列举li
+
、na
+
、k
+
、ca
2+
、mg
2+
、[fe(c5h5)2]
+
(二茂铁基团)等。另外l部分还可以为有机胺,此时l部分可以以n(r')3表示,各r'独立地选自h、c
1-6
直链或支链烷基和c
6-12
芳基,但不同时为h;或者两个r'和n可以彼此键合一起形成任选被c
1-6
直链或支链烷基取代的5-7元含氮杂环,优选为各r'独立地选自h、c
1-4
直链或支链烷基和苯基,但不同时为h;或者两个r'和n可以彼此键合一起形成任选被c
1-4
直链或支链烷基取代的5-7元的含氮的杂芳环或杂环烃。例如,可以列举甲胺、乙胺、丙胺、丁胺、二甲胺、二乙胺、二丙胺、二丁胺、三甲胺、三乙胺、三丙胺、三丁胺、n,n-二甲基苯胺、n,n-二乙基苯胺、咪唑、1-丁基-3-甲基咪唑、吡啶、哌啶等。[l]
+
部分还可以是携带碳正离子的基团,例如三苯甲基碳正离子。
[0071]
作为硼酸盐的具体例,可以列举三甲基铵四苯基硼酸盐、三乙基铵四苯基硼酸盐、三丙基铵四苯基硼酸盐、三丁基铵四苯基硼酸盐、三甲基铵四(对甲苯基)硼酸盐、三丙基铵四(对甲苯基)硼、三甲基铵四(邻,对二甲基苯基)硼酸盐、三乙基铵四(邻,对二甲基苯基)
硼酸盐、三甲基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(对三氟甲基苯基)硼酸盐、三丁基铵四(五氟苯基)硼酸盐、n,n-二乙基苯铵四苯基硼酸盐、n,n-二甲基苯铵四(五氟苯基)硼酸盐、n,n-二乙基苯铵四五氟苯基硼酸盐、二乙基铵四五氟苯基硼酸盐、三苯甲基四(五氟苯基)硼酸盐、1-丁基-3-甲基咪唑四氟硼酸盐、二茂铁四氟硼酸盐。
[0072]
本发明中,作为所述烷基铝,可以举出如下式(d)所示的化合物:al(r
11
)3ꢀꢀꢀꢀꢀ(d)在该式(d)中,基团r
11
彼此相同或不同,各自独立地选自c
1-8
烷基,优选选自甲基、乙基、丙基、异丙基、丁基、异丁基、戊基、异戊基、己基和异己基。
[0073]
在本发明的一个实施方式中,作为所述烷基铝,优选三甲基铝(al(ch3)3)、三乙基铝(al(ch2ch3)3)、三正丙基铝(al(c3h7)3)、三异丁基铝(al(i-c4h9)3)、三正丁基铝(al(c4h9)3)、三异戊基铝(al(i-c5h
11
)3)、三正戊基铝(al(c5h
11
)3)、三正己基铝(al(c6h
13
)3)、三异己基铝(al(i-c6h
13
)3)、二乙基甲基铝(al(ch3)(ch3ch2)2)和二甲基乙基铝(al(ch3ch2)(ch3)2)等,更优选三甲基铝、三乙基铝、三正丙基铝和三异丁基铝,进一步优选三乙基铝和三异丁基铝,并且最优选三异丁基铝。
[0074]
在本发明的一个实施方式中,这些烷基铝可以单独使用一种,或者以任意的比例组合使用多种。
[0075]
本发明中,作为所述烷基铝水解物,比如可以举出使前述的烷基铝与水反应后获得的水解产物。在该反应中,所述烷基铝与水的摩尔比一般为0.5-4:1,优选1-3:1。
[0076]
在本发明的一个实施方式中,这些烷基铝水解物可以单独使用一种,或者以任意的比例组合使用多种。
[0077]
本发明中,作为所述卤代烷基铝,比如可以举出下式(e)所示的化合物:al(r
11
)nx
3-n
ꢀꢀꢀꢀꢀ(e)在该式(e)中,基团r
11
彼此相同或不同,各自独立地选自c
1-8
烷基,其中优选选自甲基、乙基、丙基、异丙基、丁基、异丁基、戊基、异戊基、己基和异己基,最优选甲基或乙基;基团x为卤素,例如为氟、氯、溴、碘,优选为氯。n为1或2的整数。
[0078]
在本发明的一个实施方式中,作为所述卤代烷基铝,比如可以具体举出一氯二甲基铝(al(ch3)2cl)、二氯甲基铝(al(ch3)cl2))、一氯二乙基铝(al(ch3ch2)2cl)、二氯乙基铝(al(ch3ch2)cl2)、一氯二丙基铝(al(c3h7)2cl)、二氯丙基铝(al(c3h7)cl2))、一氯二正丁基铝(al(c4h9)2cl)、二氯正丁基铝(al(c4h9)cl2)、一氯二异丁基铝(al(i-c4h9)2cl)、二氯异丁基铝(al(i-c4h9)cl2)、一氯二正戊基铝(al(c5h
11
)2cl)、二氯正戊基铝(al(c5h
11
)cl2)、一氯二异戊基铝(al(i-c5h
11
)2cl)、二氯异戊基铝(al(i-c5h
11
)cl2)、一氯二正己基铝(al(c6h
13
)2cl)、二氯正己基铝(al(c6h
13
)cl2)、一氯二异己基铝(al(i-c6h
13
)2cl)、二氯异己基铝(al(i-c6h
13
)cl2)、一氯甲基乙基铝(al(ch3)(ch3ch2)cl)、一氯甲基丙基铝(al(ch3)(c3h7)cl)、一氯甲基正丁基铝(al(ch3)(c4h9)cl)、一氯甲基异丁基铝(al(ch3)(i-c4h9)cl)、一氯乙基丙基铝(al(ch2ch3)(c3h7)cl)、一氯乙基正丁基铝(al(ch2ch3)(c4h9)cl)和一氯乙基异丁基铝(al(ch2ch3)(i-c4h9)cl)等,其中优选一氯二乙基铝、二氯乙基铝、一氯二正丁基铝、二氯正丁基铝、一氯二异丁基铝、二氯异丁基铝、一氯二正己基铝、二氯正己基铝,进一步优选一氯二乙基铝、二氯乙基铝和一氯二正己基铝,并且最优选一氯二乙基铝。
[0079]
在本发明的一个实施方式中,这些卤代烷基铝可以单独使用一种,或者以任意的
比例组合使用多种。
[0080]
在本发明的一个实施方式中,本发明的α-烯烃-环烯烃的制备方法中,可以独立地制备包含非茂金属配合物和助催化剂的催化体系,而后将其用于α-烯烃和环烯烃的共聚;也可以在进行共聚反应时,向反应体系中依次加入非茂金属配合物和助催化剂。加入非茂金属配合物、助催化剂顺序没有特别限定。另外,对于催化剂中的有机硼化合物、选自烷基铝、烷基铝水解物、卤代烷基铝中的至少一种的烷基铝衍生物的添加顺序也没有特别限定。
[0081]
在本发明的一个实施方式中,非茂金属配合物、有机硼化合物和烷基铝衍生物可以直接使用,也可以各自配制成溶液后使用。本发明中,“直接使用”是指各化合物直接加入到用于制备上述催化体系的溶剂中;或者直接加入到进行共聚反应时的反应体系中的溶剂中。优选的是,各化合物各自配制成溶液后使用。
[0082]
作为制备催化体系的溶剂和/或配制成溶液时所使用的溶剂,可以使用本领域中已知的各种惰性有机溶剂,例如可以举出c
5-20
链烷烃、c
6-30
芳香烃、c
5-30
脂环烃、c
1-20
卤代烷烃、c
3-20
卤代脂环烃和c
6-30
卤代芳香烃,比如可以举出戊烷、己烷、庚烷、辛烷、壬烷、癸烷、十一烷、十二烷、环戊烷、环己烷、环庚烷、环辛烷、氯代戊烷、氯代己烷、氯代庚烷、氯代辛烷、氯代壬烷、氯代癸烷、氯代十一烷、氯代十二烷、氯代环己烷、甲苯、二甲苯、氯苯、二氯甲苯等,其中优选戊烷、己烷、癸烷、环己烷、甲苯、二甲苯。这些溶剂可以单独使用一种,或者以任意的比例组合使用多种。
[0083]
在本发明的一个实施方式中,在独立地制备催化体系时,可以使所述非茂金属配合物与所述助催化剂接触60-360分钟(以下称为接触反应),由此获得所述催化体系。
[0084]
在本发明的一个实施方式中,在独立地制备催化体系时,为了使非茂金属配合物、有机硼化合物和烷基铝衍生物充分接触,可以对含有非茂金属配合物、有机硼化合物和烷基铝衍生物的溶液进行搅拌(比如搅拌转速为100-1000rpm)。在本发明的一个实施方式中,在独立地制备催化体系的情况下,使得催化体系中的非茂金属配合物、有机硼化合物和烷基铝衍生物的含量比如下,或者在进行共聚反应的情况下,在反应体系中添加非茂金属配合物、有机硼化合物和烷基铝衍生物后,使各化合物的含量比如下:以al计的所述烷基铝衍生物与以金属元素m计的所述非茂金属配合物的摩尔比为50~5000:1,优选100~4000:1,更优选500~2000:1;以b计的所述有机硼化合物与以金属元素m计的所述非茂金属配合物的摩尔比为0.1~20:1,优选0.5~10:1,更优选0.8~5:1。
[0085]
需要说明的是,在本说明书中,如无特别说明,非茂金属配合物的摩尔量通常以金属元素m计,烷基铝衍生物的摩尔量通常以金属元素al计,有机硼化合物的摩尔量通常以元素b计。
[0086]
在本发明的一个实施方式中,对所述共聚方法的反应方式没有特别的限定,可以采用本领域公知的那些,比如可以举出溶液法、本体法等,其中优选溶液法。
[0087]
在本发明的一个实施方式中,所述共聚方法根据需要可以在惰性有机溶剂作为共聚用溶剂的存在下进行。作为所述共聚用溶剂,可以举出本领域在进行链烯烃与环烯烃的共聚时常规使用的那些,用量也可以参考现有技术的常规用量,并没有特别的限定。具体举例而言,作为所述共聚用溶剂,比如可以举出c
5-20
链烷烃、c
6-30
芳香烃、c
5-20
脂环烃、c
1-20
卤代链烷烃、c
3-20
卤代脂环烃和c
6-30
卤代芳香烃等,其中优选c
5-12
直链或支链链烷烃、c
5-12
环烷烃、c
6-12
芳烃、c
1-12
直链或支链卤代链烷烃、c
3-12
卤代环烷烃和c
6-12
卤代芳烃,更优选c
6-9
直链或支链链烷烃、c
6-9
环烷烃、c
6-10
芳烃、c
1-8
直链或支链卤代烷烃、c
3-8
卤代环烷烃和c
6-10
卤代芳烃,最优选戊烷、己烷、庚烷、环己烷、环辛烷、甲苯或者二甲苯。这些共聚用溶剂可以单独使用一种,或者以任意的比例组合使用多种。
[0088]
在本发明的一个实施方式中,所述共聚反应可在带搅拌的塔式或釜式反应器中进行,优选釜式反应器。反应器容积为0.05-1000l,优选0.1-100l。
[0089]
在本发明的一个实施方式中,所述共聚方法的反应压力(总压)为0.1-5.0mpa,优选0.1-3.0mpa,更优选0.1-2.0mpa,但并不限于此。
[0090]
在本发明的一个实施方式中,所述共聚方法的反应温度为40-100℃,优选60-100℃,更优选70-90℃,但并不限于此。
[0091]
在本发明的一个实施方式中,进行共聚反应时,反应体系中的环烯烃与以金属元素m计的非茂金属配合物的摩尔比为10
5-107:1,优选5
×
10
5-5
×
106:1,更优选7
×
10
5-2
×
106:1,但并不限于此。
[0092]
本发明的共聚方法中,聚合时间与催化剂的用量和反应温度有关,催化剂的用量越多,反应温度越高反应速率越快,反应时间越短,反应时间一般为0.1-10h,优选0.1-5h,更优选15min-2h,但并不限于此。
[0093]
在本发明的一个实施方式中,在进行共聚反应时,可以在催化体系的基础上,进一步添加链转移剂,调节共聚物的重均分子量、分子量分布、环烯烃单元的含量等。作为所述链转移剂,可以列举出本发明助催化剂之外的其他金属烷基化合物,例如,可以列举正丁基锂、二乙基锌、二丙基锌、二丁基锌、二异丁基锌、二乙基镁、二丁基镁或正丁基乙基镁中的一种或多种。以金属元素计的所述链转移剂与以金属元素m计的所述非茂金属配合物的摩尔比为5~500:1,优选10~100:1,更优选10~50:1。在使用链转移剂时,可以在独立制备的催化剂体系中添加链转移剂,也可以在进行共聚反应时,向反应体系中添加非茂金属配合物、助催化剂时添加链转移剂。另外,链转移剂的添加顺序没有特别限定。链转移剂可以直接使用,也可以制备成溶液后使用。制备溶液时,可以采用本发明上述的用于制备非茂金属配合物、有机硼化合物、烷基铝衍生物等溶液时所列举的溶剂。
[0094]
需要说明的是,在本说明书中,如无特别说明,链转移剂的摩尔量都是以链转移剂中的金属元素的摩尔量计,例如,当以二乙基锌作为链转移剂时,链转移剂的摩尔量以锌计。
[0095]
在本发明的一个实施方式中,可采用单聚合反应器装置以连续或间歇聚合方式,在单一反应器中制备出α-烯烃-环烯烃共聚物。
[0096]
在本发明的一个实施方式中,本发明的α-烯烃和环烯烃的共聚方法例如可以如下进行:在温度为40-100℃、压力为0.1-5.0mpa条件下,将共聚用溶剂、α-烯烃及环烯烃加入反应器;待α-烯烃和环烯烃在共聚用溶剂中溶解至饱和后,依次加入非茂金属配合物溶液、烷基铝衍生物溶液、有机硼化合物溶液、和任选的链转移剂,使得环烯烃与以金属元素m计的非茂金属配合物的摩尔量之比为10
5-107:1,以al计的所述烷基铝衍生物与以金属元素m计的所述非茂金属配合物的摩尔比为50~5000:1,以b计的所述有机硼化合物与以金属元素m计的所述非茂金属配合物的摩尔比达到0.1~20:1,以金属元素计的所述链转移剂与以金属元素m计的所述非茂金属配合物的摩尔比为5~500:1;反应过程中为保持反应器内压力而通入α-烯烃,保温聚合反应时间为15min~2h,由此,可以制备得到α-烯烃-环烯烃共聚
物。
[0097]
在本发明的一个实施方式中,反应结束后将所得共聚产物在酸化乙醇中进行沉淀。具体而言,将所得聚合后溶液倒入酸化乙醇中沉淀后烘干,优选在60℃下抽真空烘干24h,由此可以得到纯化后的α-烯烃-环烯烃共聚物。所述酸化乙醇由浓盐酸和乙醇混合后制备,所述浓盐酸相对于乙醇的体积百分比可以为1v/v%-25v/v%,还可以为10v/v%-20v/v%,例如可以为15v/v%。
[0098]
在本发明的一个实施方式中,在共聚反应时,反应体系中以金属元素m计的所述非茂金属配合物在反应溶液中的浓度为0.1
×
10-5
mol/l~50
×
10-5
mol/l,优选0.5
×
10-5
mol/l~20
×
10-5
mol/l,更优选1
×
10-5
mol/l~10
×
10-5
mol/l。
[0099]
在本发明的一个实施方式中,在共聚反应时,反应体系中所述环烯烃在反应溶液中的浓度为1~100mol/l,优选为10~50mol/l。
[0100]
在本发明的一个实施方式中,在共聚反应时,反应体系中以b计的所述有机硼化合物在反应中溶液的浓度为1
×
10-6
mol/l~100
×
10-5
mol/l,优选为5
×
10-6
mol/l~50
×
10-5
mol/l。
[0101]
在本发明的一个实施方式中,在共聚反应时,反应体系中以al计的所述烷基铝衍生物在反应中溶液的浓度为1
×
10-3
mol/l~200
×
10-3
mol/l,优选10
×
10-3
mol/l~150
×
10-3
mol/l。
[0102]
在本发明的一个实施方式中,在共聚反应时,反应体系中以金属元素计的所述链转移剂在反应中溶液的浓度为0~500
×
10-5
mol/l,优选10
×
10-5
mol/l~300
×
10-5
mol/l。
[0103]
在本发明的一个实施方式中,所述共聚方法可以在有氢气存在的条件下进行,也可以在没有氢气存在的条件下进行。在氢气存在的情况下,氢气的分压可以是前述反应压力的0.01-99%,优选0.01-50%,但有时并不限于此。
[0104]
在本发明的一个实施方式中,所述共聚方法可以在有非活性气体存在的条件下进行,也可以在没有非活性气体存在的条件下进行。在非活性气体存在的情况下,非活性气体的分压可以是前述反应压力的0.01-99%,优选0.01-50%,但有时并不限于此。作为所述非活性气体,比如可以举出氮气、氦气或者氩气等。这些非活性气体可以单独使用一种,也可以根据需要以任意的比例组合使用多种。
[0105]
本发明还提供一种聚合物组合物,其至少包含本发明的α-烯烃-环烯烃共聚物。
[0106]
在本发明中的聚合物组合物中,还可以在不损害本发明目的的范围内,根据需要进一步添加本领域公知的其他的添加剂,例如加工热稳定剂、光稳定剂、紫外线吸收剂、抗氧化剂、着色剂、抗静电剂、阻燃剂、拒水剂、防水剂、亲水性赋予剂、导电性赋予剂、导热性赋予剂、电磁屏蔽性赋予剂、透光性调节剂、荧光剂、滑动性赋予剂、透明性赋予剂、抗结块剂、金属减活剂、抗菌剂、填料等,但不限于此。
[0107]
本发明还涉及α-烯烃-环烯烃共聚物用于制造所述聚合物组合物的应用。
[0108]
在制造光学部件、包装材料、电子部件、医疗器具时,可以根据需要采用本领域常用的技术,例如,可以举出挤出成型、注射成型、压延成型、吹塑成型和热成型等成型方法,但不限于此。由此,通过这些成型方法,利用本发明的α-烯烃-环烯烃共聚物或者本发明的聚合物组合物来制造光学部件、包装材料、电子部件、医疗器具。
[0109]
因此,本发明还提供所述α-烯烃-环烯烃共聚物在制造光学部件、包装材料、电子
部件、医疗器具中的应用。本发明还提供所述聚合物组合物在制造光学部件、包装材料、电子部件、医疗器具中的应用。
实施例
[0110]
以下采用实施例进一步详细地说明本发明,但本发明并不限于这些实施例。
[0111]
以下实施例中,共聚物中环烯烃含量用核磁共振仪测定。
[0112]
具体而言,如图1所示,以降冰片烯为例,用avance iii hd核磁共振仪测定样品中的降冰片烯单元的含量(nb%),测试温度为120℃。共聚物样品用氘代邻二氯苯溶解配成约20wt%的溶液,在120℃下扫描6000次得到样品的
13
c nmr谱图。图中的主要核磁共振特征信号峰可归结为以下四组:45~55ppm之间的信号归属于c2/c3,37~44ppm之间的信号归属于c1/c4,32~36ppm之间的信号归属于c7,31.5ppm以下归属于c5/c6和乙烯单元的亚甲基信号。共聚物中降冰片烯单元的含量根据
13
c-nmr谱图中各归属信号峰的峰面积计算而得到:。
[0113]
mη分子量和pdi的测定方法共聚物分子量及其分布用polymer laboratories的pl-220型凝胶渗透色谱仪测试,以1,2,4-三氯苯为流动相,聚苯乙烯为标样,采用示差检测器,流速为1.0ml/min,测定温度为150℃,样品浓度为2.0mg/ml。
[0114]
共聚物的玻璃化转变温度的测定。
[0115]
共聚物的热性能采用差式扫描量热仪(perkin-elmer dsc 7)测定。测试的样品量为3-5mg,气氛为氮气。样品首先以20℃/min的速率由30℃加热到180℃,停留3min,再以20℃/min的速率降至20℃,停留3min,然后以20℃/min的速率再升至180℃。使用第二次升温曲线进行分析。
[0116]
实施例1:在1l不锈钢釜式反应器中,将15.6mol精制降冰片烯溶于500ml精制甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在90℃、1mpa及搅拌的条件下,一次加入30ml浓度为1mol/l三异丁基铝的甲苯溶液、40μmol三苯甲基四(五氟苯基)硼酸盐、20μmol 3-叔丁基亚水杨基2-甲硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0117]
反应15min后,产生凝胶,将反应液倒入含15%(v/v%)盐酸的乙醇(由300ml盐酸和2000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(1000ml)洗涤后,干燥(60℃干燥至恒重),得共聚物14克,催化剂活性2.8
×
106g/(molm
·
h)。共聚物物性见表1。
[0118]
实施例2:本实施例使用的是2l釜式反应器。将24mol精制乙叉降冰片烯溶于1000ml精制环己烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在55℃、1mpa及搅拌的条件下,一次加入80ml浓度为1mol/l三正丁基铝的甲苯溶液、0.8ml浓度为1mol/l二乙基锌甲苯溶液、120μmol n,n-二甲基苯铵四(五氟苯基)硼酸盐、60μmol 3,5-二叔丁基水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0119]
反应0.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由450ml盐酸和3000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(1000ml)洗涤后,干燥(60℃干燥至恒重),得共聚物30g,催化剂活性1.0
×
106g/(molm
·
h)。共聚物物性见表1。
[0120]
实施例3:本实施例使用的是2l釜式反应器。将30mol精制乙烯基降冰片烯溶于1000ml精制二甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在75℃、1mpa及搅拌的条件下,一次加入40ml浓度为1mol/l三异戊基铝的甲苯溶液、2ml浓度为1mol/l二乙基锌甲苯溶液、40μmol 1-丁基-3-甲基咪唑四氟硼酸盐、40μmol水杨基2-甲硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0121]
反应1h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由600ml盐酸和4000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物48g,催化剂活性1.2
×
106g/(molm
·
h)。共聚物物性见表1。
[0122]
实施例4:本实施例使用的是1l釜式反应器。将16mol精制降冰片烯溶于500ml精制二甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在70℃、1mpa及搅拌的条件下,一次加入10ml浓度为1mol/l三正己基铝的甲苯溶液、0.4ml浓度为1mol/l二乙基锌甲苯溶液、20μmol二茂铁四氟硼酸盐、20μmol亚水杨基2-苯硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0123]
反应15min后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物7g,催化剂活性1.4
×
106g/(molm
·
h)。共聚物物性见表1。
[0124]
实施例5:在5l不锈钢釜式反应器中,将90mol精制乙烯基降冰片烯溶于2500ml精制环辛烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在65℃、2mpa及搅拌的条件下,一次加入50ml浓度为1mol/l三异己基铝的甲苯溶液、200μmol三苯甲基四(五氟苯基)硼酸盐、2ml浓度为1mol/l二乙基锌甲苯溶液、200μmol亚水杨基2-苯硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0125]
反应2h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由750ml盐酸和5l乙醇组成)中沉淀后过滤,滤饼用乙醇(2l)洗涤后,干燥(60℃干燥至恒重),得共聚物600克,催化剂活性1.5
×
106g/(molm
·
h)。共聚物物性见表1。
[0126]
实施例6:本实施例使用的是1l釜式反应器。将21mol精制乙叉降冰片烯溶于500ml精制甲苯、二甲苯混合溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在80℃、2mpa及搅拌的条件下,一次加入20ml浓度为1mol/l二乙基甲基铝的甲苯溶液、60μmol n,n-二甲基苯铵四(五氟苯基)硼酸盐、0.4ml浓度为1mol/l二乙基锌甲苯溶液、20μmol 3,5-二叔丁基水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0127]
反应0.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共
聚物11g,催化剂活性1.1
×
106g/(molm
·
h)。共聚物物性见表1。
[0128]
实施例7:本实施例使用的是0.5l釜式反应器。将9mol精制乙叉降冰片烯溶于250ml精制甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在50℃、2mpa及搅拌的条件下,一次加入15ml浓度为1mol/l二甲基乙基铝的甲苯溶液、0.2ml浓度为1mol/l二乙基锌甲苯溶液、10μmol 1-丁基-3-甲基咪唑四氟硼酸盐、10μmol 3-叔丁基亚水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0129]
反应0.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物14g,催化剂活性2.8
×
106g/(molm
·
h)。共聚物物性见表1。
[0130]
实施例8:本实施例使用的是0.5l釜式反应器。将10mol精制乙烯基降冰片烯溶于300ml精制环辛烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在40℃、2mpa及搅拌的条件下,一次加入40ml浓度为1mol/l三正丙基铝的甲苯溶液、1ml浓度为1mol/l二乙基锌甲苯溶液、20μmol二茂铁四氟硼酸盐、10μmol 3,5-二叔丁基亚水杨基2-巯基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0131]
反应1.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物12g,催化剂活性0.8
×
106g/(molm
·
h)。共聚物物性见表1。
[0132]
实施例9:在1l不锈钢釜式反应器中,将13.5mol精制降冰片烯溶于500ml精制甲苯、环己烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1.5mpa),使溶液被乙烯饱和,在55℃、1.5mpa及搅拌的条件下,一次加入5ml浓度为1mol/l三异己基铝的甲苯溶液、5μmol三苯甲基四(五氟苯基)硼酸盐、0.2ml浓度为1mol/l二乙基锌甲苯溶液、10μmol 3-叔丁基亚水杨基2-甲硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1.5mpa。
[0133]
反应15min后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物5克,催化剂活性2
×
106g/(molm
·
h)。共聚物物性见表1。
[0134]
实施例10:本实施例使用的是1l釜式反应器。将18mol精制乙烯基降冰片烯溶于500ml精制甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(3mpa),使溶液被乙烯饱和,在95℃、3mpa及搅拌的条件下,一次加入20ml浓度为1mol/l三异丁基铝的甲苯溶液、1ml浓度为1mol/l二乙基锌甲苯溶液、40μmol n,n-二甲基苯铵四(五氟苯基)硼酸盐、20μmol 3,5-二叔丁基水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为3mpa。
[0135]
反应15min后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由300ml盐酸和2000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共
聚物15g,催化剂活性3
×
106g/(molm
·
h)。共聚物物性见表1。
[0136]
实施例11:本实施例使用的是10l釜式反应器。将225mol精制乙叉降冰片烯溶于5l精制二甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(3mpa),使溶液被乙烯饱和,在40℃、3mpa及搅拌的条件下,一次加入200ml浓度为1mol/l三异丁基铝的甲苯溶液、10ml浓度为1mol/l二乙基锌甲苯溶液、800μmol 1-丁基-3-甲基咪唑四氟硼酸盐、200μmol 3,5-二叔丁基亚水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为3mpa。
[0137]
反应1h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由1.5l盐酸和10l乙醇组成)中沉淀后过滤,滤饼用乙醇(3l)洗涤后,干燥(60℃干燥至恒重),得共聚物52g,催化剂活性0.26
×
106g/(molm
·
h)。共聚物物性见表1。
[0138]
实施例12:本实施例使用的是2.5l釜式反应器。将54mol精制降冰片烯溶于1.5l精制环己烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(3mpa),使溶液被乙烯饱和,在85℃、1mpa及搅拌的条件下,一次加入80ml浓度为1mol/l三异戊基铝的甲苯溶液、0.8ml浓度为1mol/l二乙基锌甲苯溶液、160μmol二茂铁四氟硼酸盐、50μmol 3,5-二叔丁基亚水杨基2-巯基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为3mpa。
[0139]
反应1h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由3000ml盐酸和20l乙醇组成)中沉淀后过滤,滤饼用乙醇(3l)洗涤后,干燥(60℃干燥至恒重),得共聚物200g,催化剂活性4
×
106g/(molm
·
h)。共聚物物性见表1。
[0140]
比较例1:本实施例使用的是1l釜式反应器。将16mol精制降冰片烯溶于500ml精制二甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在70℃、1mpa及搅拌的条件下,一次加入0.4ml浓度为1mol/l二乙基锌甲苯溶液、20ml浓度为1.5mol/l改性甲基铝氧烷的甲苯溶液、20μmol亚水杨基2-苯硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0141]
反应15min后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物8g,催化剂活性1.6
×
106g/(molm
·
h)。共聚物物性见表1。
[0142]
比较例2:在5l不锈钢釜式反应器中,将90mol精制乙烯基降冰片烯溶于2500ml精制环辛烷配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在65℃、2mpa及搅拌的条件下,一次加入30ml浓度为1.9mol/l甲基铝氧烷的甲苯溶液,2ml浓度为1mol/l二乙基锌甲苯溶液、200μmol亚水杨基2-苯硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0143]
反应2h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由750ml盐酸和5l乙醇组成)中沉淀后过滤,滤饼用乙醇(2l)洗涤后,干燥(60℃干燥至恒重),得共聚物675克,催化剂活性1.7
×
106g/(molm
·
h)。共聚物物性见表1。
[0144]
比较例3:
本实施例使用的是1l釜式反应器。将21mol精制乙叉降冰片烯溶于500ml精制甲苯、二甲苯混合溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(2mpa),使溶液被乙烯饱和,在80℃、2mpa及搅拌的条件下,一次加入20ml浓度为1.9mol/l甲基铝氧烷的甲苯溶液、0.4ml浓度为1mol/l二乙基锌甲苯溶液、20μmol 3,5-二叔丁基水杨基2-丙硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为2mpa。
[0145]
反应0.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由150ml盐酸和1000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物10g,催化剂活性1.0
×
106g/(molm
·
h)。共聚物物性见表1。
[0146]
比较例4:在1l不锈钢釜式反应器中,将15.6mol精制降冰片烯溶于200ml精制甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(1mpa),使溶液被乙烯饱和,在90℃、1mpa及搅拌的条件下,一次加入10ml浓度为1.9mol/l甲基铝氧烷的甲苯溶液、10μmol 3-叔丁基亚水杨基2-甲硫基苯胺三氯化钛,聚合过程通过补充计量加入乙烯,控制压力为1mpa。
[0147]
反应15min后,将反应液倒入含15%(v/v%)盐酸的乙醇(由300ml盐酸和2000ml乙醇组成)中沉淀后过滤,滤饼用乙醇(1000ml)洗涤后,干燥(60℃干燥至恒重),得共聚物6克,催化剂活性1.2
×
106g/(molm
·
h)。共聚物物性见表1。
[0148]
比较例5在250ml玻璃反应器中,将0.013mol精制降冰片烯溶于50ml精制甲苯配成溶液,将该溶液加入到预先用氮气置换过的反应器中,多次冲压乙烯(0.1mpa),使溶液被乙烯饱和,在70℃、0.1mpa及搅拌的条件下,依次加入1.03ml浓度为1.94mol/l改性甲基铝氧烷(mmao)的甲基环己烷溶液、2μmol rac-乙烯基双(4,5,6,7四氢-1-茚基)二氯化锆,控制压力为0.1mpa。
[0149]
反应0.5h后,停止反应,将反应液倒入含15%(v/v%)盐酸的乙醇(由15ml盐酸和100ml乙醇组成)中沉淀后过滤,滤饼用乙醇(300ml)洗涤后,干燥(60℃干燥至恒重),得共聚物5.37克,催化剂活性5.37
×
106g/(mol
·
h)。共聚物物性见表1。
[0150]
表1 催化剂活性以及共聚物物性
[0151]
通过本发明的实施例可以证实,在不使用铝氧烷系助催化剂的情况下,通过使用包含烷基铝衍生物和有机硼化合物作为助催化剂的非茂金属催化体系,可以低成本且高效地制备α-烯烃-环烯烃共聚物。共聚反应的活性与使用铝氧烷系助催化剂的情况相当甚至
更为优异,并且所得共聚物的物性也相当甚至更为优异。特别是,与使用茂金属催化体系的情况相比,本发明的非茂金属催化体系表现出更为优异的活性,所得共聚物的物性也更为优异。
[0152]
最后,还需要注意的是,以上列举的仅是本发明的若干个具体实施例。显然,本发明不限于以上实施例,还可以有许多变形。本领域的普通技术人员能从本发明公开的内容直接导出或联想到的所有变形,均应认为是本发明的保护范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1