一种苯偶酰类化合物及其制备方法和应用
1.本发明属于化学技术领域,具体涉及一种苯偶酰类化合物及其制备方法和应用,尤其涉及一种检测效果好的苯偶酰类化合物及其制备方法和应用。
背景技术:
2.胍类化合物广泛存在于人体中,参与体内各种代谢循环、维持人体内环境的稳定,是重要的具有生物活性的小分子化合物。例如精氨酸、高精氨酸、肌酸、肌酐等在尿素循环中扮演着重要的角色。由精氨酸甲基转化酶i、ii、iii型催化精氨酸的胍基部分发生甲基化形成的甲基化精氨酸,参与多种生物代谢过程,其中最重要的是非对称性二甲基精氨酸,它是一氧化氮合成酶的抑制剂,在调节血管张力和内皮细胞功能方面起着重要作用,血管中非对称性二甲基精氨酸的改变会引起一系列心血管疾病。内源性胍基丁胺是经精氨酸脱羧酶脱去精氨酸的羧基后的产物,它广泛的分布于众多器官和组织中,具有器官特异性。迄今为止,已发现胍基丁胺在中枢神经系统、心血管系统等系统的体内代谢中存在多种生理功能。在肝、肾、胰等器官中,由甘氨酸和精氨酸在酶的作用下形成的胍乙酸,广泛分布在身体的各个组织器官,是人体合成肌酸的主要内源性物质,对人体能量代谢有着重要影响。由此可见,对胍类化合物进行检测分析具有重要的生理学和病理学意义,但由于许多胍类化合物其微弱的含量,以及其较低的分子量,使得对胍类化合物的检测存在较大的干扰,同时也造成其定性定量分析困难。
3.cn104076036b公开了一种检测制品中是否添加有双胍类物质的方法,该方法包括以下步骤:(1)取待测的制品适量,与提取溶剂混合,振摇使充分混合,得混合试液;(2)将步骤(1)所得混合试液过滤,得澄清滤液;(3)向步骤(2)所得滤液中加入检测试剂,摇匀,得混合液,目视观察混合液的变化;(4)如果步骤(3)所得混合液出现沉淀,则表示所述制品中添加有双胍类物质;如果所述混合液未出现沉淀,则表示所述制品中未添加有双胍类物质。该发明方法与现有方法相比,成本低、操作过程简单,适用于在基层部门广泛开展。但其只能定性监测双胍类化合物,未涉及单胍类化合物的定性定量检测。
4.目前对胍类化合物的分析方法专属性差、检测灵敏度较低、检测通量低,同时已知的方法只能针对已知的胍类化合物进行分析检测,无法满足实际样本中未知胍的检测。因此,如何提供一种检测灵敏度高、检测效果好的胍类化合物检测方法,成为了亟待解决的问题。
技术实现要素:
5.针对现有技术的不足,本发明的目的在于提供一种苯偶酰类化合物及其制备方法和应用,尤其提供一种检测效果好的苯偶酰类化合物及其制备方法和应用。本发明提供的苯偶酰类化合物应用在胍类化合物的检测中,能够显著提高检测的灵敏度、准确性、稳定性,同时能够用于对待测样本中潜在胍类化合物的识别,成本低。
6.为达到此发明目的,本发明采用以下技术方案:
7.第一方面,本发明提供了一种苯偶酰类化合物,所述苯偶酰类化合物的结构如式i所示:
[0008][0009]
其中r1为
‑
(ch2)
n
‑
n(cx1y1z1)(cx2y2z2),n为1
‑
6的整数,x1、x2、y1、y2、z1、z2独立地选自氢或氘,其中,n可以是1、2、3、4、5或6。
[0010]
上述特定结构的化合物应用在胍类化合物的检测中,对待测样本中的胍类化合物进行特异性标记衍生化,改善了胍类化合物原型在lc(液相色谱)分析过程中色谱行为差的缺点,在ms(质谱)分析过程中产物以双电荷离子形式存在,二级碎裂后会产生一系列特征碎片离子,具有很好的灵敏度、很强的专属性及选择性,能显著提高定性定量分析的准确性,同时稳定性好,能够用于对待测样本中潜在胍类化合物的识别,成本低。
[0011]
第二方面,本发明提供了如上所述的苯偶酰类化合物的制备方法,所述制备方法包括以下步骤:将化合物a、甲醛或氘代甲醛以及甲酸或氘代甲酸反应,得到所述苯偶酰类化合物,化合物a的结构如式ii所示:
[0012][0013]
其中r2选自
‑
(ch2)
n
‑
nh2或
‑
(ch2)
n
‑
nhcx3,n具有与上述相同的限定范围,x为氢或氘。
[0014]
上述制备方法能够快速方便制备所述苯偶酰类化合物。
[0015]
优选地,所述r2选自
‑
(ch2)
n
‑
nh2、n为2
‑
6的整数,例如2、3、4、5或6,所述化合物a由包括以下步骤的制备方法制备得到:
[0016]
(1)将化合物a1与邻苯二甲酸酐反应,得到化合物b1;
[0017]
(2)将步骤(1)得到的化合物b1与碘、三苯基膦反应,得到化合物c1;
[0018]
(3)将步骤(2)得到的化合物c1与4,4'
‑
二溴联苯甲酰反应,得到所述化合物a。
[0019]
反应式如式iii所示:
[0020][0021]
其中m为1
‑
5的整数,例如1、2、3、4或5。
[0022]
优选地,所述r2选自
‑
(ch2)
n
‑
nh2、n为1,所述化合物a由包括以下步骤的制备方法
制备得到:将4,4'
‑
二甲基联苯甲酰与nbs(n
‑
溴代丁二酰亚胺)反应,之后将产物与nah(氢化钠)、bocnh2(氨基甲酸叔丁酯)反应,经盐酸处理得到所述化合物a。
[0023]
优选地,r2选自
‑
(ch2)
n
‑
nhcx3、n为2
‑
6的整数,例如2、3、4、5或6,x为氢或氘,所述化合物a由包括以下步骤的制备方法制备得到:
[0024]
(1’)将化合物a2与二碳酸二叔丁酯反应,得到化合物b2;
[0025]
(2’)将步骤(1’)得到的化合物b2与tbscl(叔丁基二甲基氯硅烷)反应,得到化合物c2;
[0026]
(3’)将步骤(2’)得到的化合物c2与ch3i(碘甲烷)或cd3i(全氘代碘甲烷)反应,得到化合物d;
[0027]
(4’)将步骤(3’)得到的化合物d与tbaf(四丁基氟化铵)反应,之后与碘、三苯基膦反应,得到化合物e;
[0028]
(5’)将步骤(4’)得到的化合物e与4,4'
‑
二溴联苯甲酰反应,得到所述化合物a。
[0029]
反应式如式iv所示:
[0030][0031]
其中,p为1
‑
5的整数,例如1、2、3、4或5,x为氢或氘。
[0032]
优选地,r2选自
‑
(ch2)
n
‑
nhcx3、n为1、x为氢或氘,所述化合物a由包括以下步骤的制备方法制备得到:将4,4'
‑
二甲基联苯甲酰与nbs反应,得到产物甲;之后将产物甲与nah、bocnh2反应,得到产物乙;最后将产物乙、nah与ch3i或cd3i反应,经三氟乙酸处理得到所述化合物a。
[0033]
第三方面,本发明提供了如上所述的苯偶酰类化合物在胍类化合物检测中的应用。
[0034]
第四方面,本发明提供了一种胍类化合物的定性检测方法,所述定性检测方法包括以下步骤:将待测样品与如上所述的苯偶酰类化合物反应,之后将产物进行lc
‑
ms检测,根据检测结果确定待测样品中是否含有胍类化合物以及胍类化合物结构。
[0035]
第五方面,本发明提供了一种胍类化合物的相对定量检测方法,所述相对定量检测方法包括以下步骤:将如上所述的苯偶酰类化合物与待测样品反应,得到样本a;将如上所述的苯偶酰类化合物和与待测样品中含有的胍类化合物相同的胍类化合物的标准溶液反应,得到样本b;之后取样本a与样本b等体积合并,进行lc
‑
ms检测,根据检测结果确定待测样品中胍类化合物含量与胍类化合物的标准溶液中胍类化合物含量的比值实现相对定量检测。
[0036]
优选地,所述与待测样品反应的如上所述的苯偶酰类化合物以及所述和与待测样
品中含有的胍类化合物相同的胍类化合物的标准溶液反应的如上所述的苯偶酰类化合物的n相同,且结构中氘的数目不同。
[0037]
第六方面,本发明还提供了一种胍类化合物的绝对定量检测方法,所述绝对定量检测方法包括以下步骤:将待测样本与如上所述的苯偶酰类化合物反应,之后经lc
‑
ms检测,并用标准曲线计算得到待测样本中胍类化合物的含量。
[0038]
相对于现有技术,本发明具有以下有益效果:
[0039]
本发明制备得到了一种具有特定结构的苯偶酰类化合物,应用在胍类化合物的检测中,能够对待测样本中的胍类化合物进行特异性标记衍生化,改善了胍类化合物原型在lc分析过程中色谱行为差的缺点,在ms分析过程中产物以双电荷离子形式存在,二级碎裂后会产生一系列特征碎片离子,具有很好的灵敏度、很强的专属性及选择性,能显著提高定性定量分析的准确性,同时稳定性好,能够用于对待测样本中潜在胍类化合物的识别,成本低。
附图说明
[0040]
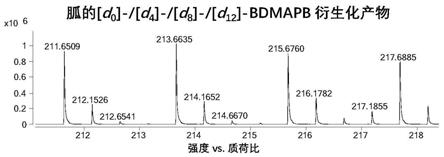
图1是标记效果测试中胍与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0041]
图2是标记效果测试中胍乙酸与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0042]
图3是标记效果测试中肌酸与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0043]
图4是标记效果测试中胍基丁酸与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0044]
图5是标记效果测试中精氨酸与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0045]
图6是标记效果测试中高精氨酸与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果;
[0046]
图7是标记效果测试中胍乙酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的lc结果;
[0047]
图8是标记效果测试中胍乙酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的二级质谱结果;
[0048]
图9是标记效果测试中肌酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的lc结果;
[0049]
图10是标记效果测试中肌酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的二级质谱结果;
[0050]
图11是胍的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的稳定性测试结果;
[0051]
图12是胍乙酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的稳定性测试结果;
[0052]
图13是肌酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物
的稳定性测试结果;
[0053]
图14是胍基丁酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb衍生化产物的稳定性测试结果;
[0054]
图15是精氨酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的稳定性测试结果;
[0055]
图16是高精氨酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的稳定性测试结果。
具体实施方式
[0056]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0057]
以下测试实验中
[0058]
制备例1化合物a(n=3,r=
‑
(ch2)
n
‑
nh2)的盐酸盐的合成
[0059]
化合物a(n=3,r=
‑
(ch2)
n
‑
nh2)的盐酸盐的结构如下所示:
[0060][0061]
制备方法如下:
[0062]
(1)向三口瓶中加入10.0g 3
‑
氨基丙醇与19.7g邻苯二甲酸酐,加入溶剂超干甲苯,在125℃下回流反应,同时利用分水器除去反应中产生的水,反应6h后,旋干反应溶剂,得化合物3
‑
(邻苯二甲酰亚氨基)丙醇。
[0063]
(2)在1l的反应烧瓶中,加入21.4g步骤(1)得到的化合物3
‑
(邻苯二甲酰亚氨基)丙醇、51.1g三苯基磷、13.3g咪唑,氮气保护后,冰浴下,加入300ml溶剂二氯甲烷,搅拌,随后分批加入49.5g的i2,25℃下反应12h,之后加入纯水,终止反应,用二氯甲烷萃取、干燥,之后柱层析,得化合物邻苯二甲酰亚氨基丙基碘,产率为72%。
[0064]
(3)称取5.2g步骤(2)得到的邻苯二甲酰亚氨基丙基碘和6.2g锌粉,置于50ml反应瓶中,加入10mg碘,氮气保护后,加入30ml溶剂dmf,60℃下搅拌1h。之后在25℃下静置沉淀,取上清液,加入氮气保护下,并称有1.9g对溴苯偶酰、171.8mg醋酸钯、277.4mg三苯基磷和10ml溶剂dmf的反应瓶中,65℃下搅拌9h。之后利用ea
‑
h2o萃取反应液,有机相利用无水硫酸钠干燥后旋干,柱层析纯化,得化合物4,4
’‑
二(邻苯二甲酰亚氨基丙基)苯偶酰产率65%。
[0065]
(4)称取100mg 4,4
’‑
二(邻苯二甲酰亚氨基丙基)苯偶酰于30ml封管中,加入10ml浓盐酸,150℃下搅拌12h。冷却至室温后,旋干溶剂,加入乙酸乙酯,滤出固体得到化合物a(n=3,r=
‑
(ch2)
n
‑
nh2)的盐酸盐,产率为85%。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.89(d,j=8.0hz,4h),7.47(d,j=8.0hz,4h),2.96(t,j=7.8hz,4h),2.83(t,j=7.9hz,4h),2.00(tt,j=9.8,6.6hz,4h)。
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.75,131.25,129.82,128.98,38.82,32.22,28.41。
[0066]
制备例2[d0]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d0]
‑
bdmapb)的合成
[0067]
化合物[d0]
‑
bdmapb的结构如下所示:
[0068][0069]
制备方法如下:在反应管中,加入100mg制备例1得到的化合物a(n=3,r=
‑
(ch2)
n
‑
nh2)的盐酸盐、20%的甲醛2ml和1ml甲酸,100℃下反应12h,旋干反应溶剂,柱层析,得到[d0]
‑
bdmapb,产率90%。其中[d0]指代结构中不包含氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.88(d,j=8.3hz,4h),7.47(d,j=8.2hz,4h),3.18
–
3.12(m,4h),2.88(s,12h),2.81(t,j=7.7hz,4h),2.12
–
2.03(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.38,131.33,129.83,129.01,56.97,42.04,32.05,25.40。
[0070]
制备例3[d8]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d8]
‑
bdmapb)的合成
[0071]
化合物[d8]
‑
bdmapb的结构如下所示:
[0072][0073]
制备方法中除用等量氘代甲酸代替甲酸外,其余与制备例2一致。其中[d8]指代结构中包含八个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.89(d,j=8.3hz,4h),7.47(d,j=8.4hz,4h),3.16
–
3.12(m,4h),2.84(s,4h),2.81(t,j=7.7hz,4h),2.11
–
2.04(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.45,148.41,131.32,129.83,129.02,56.84,41.50,32.05,25.38。
[0074]
制备例4[d
12
]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d
12
]
‑
bdmapb)的合成
[0075]
化合物[d
12
]
‑
bdmapb的结构如下所示:
[0076][0077]
制备方法中除用等量氘代甲酸代替甲酸、用等量氘代甲醛替代甲醛外,其余与制备例2一致,其中[d
12
]指代结构中包含十二个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.90(d,j=7.1hz,4h),7.47(d,j=8.1hz,4h),3.16
–
3.12(m,4h),2.82(t,j=7.7hz,4h),2.10
–
2.04(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.43,148.39,131.35,129.83,129.01,56.79,39.80,32.04,25.37。
[0078]
制备例5 n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷的合成
[0079]
n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷结构如下所示:
[0080][0081]
制备方法如下:
[0082]
(1)向三口瓶中加入11.2g 3
‑
氨基
‑1‑
丙醇与33.6g二碳酸二叔丁酯,加200ml无水二氯甲烷,在25℃反应6h,水洗,有机相干燥浓缩得化合物n
‑
(叔丁氧羰基)丙醇胺。
[0083]
(2)向反应瓶中加入16.1g步骤(1)得到的n
‑
(叔丁氧羰基)丙醇胺、16.5g叔丁基二甲基氯硅烷、10.2g咪唑、0.2g对二甲氨基吡啶和300ml二氯甲烷,搅拌反应10h,依次用饱和
氯化铵水溶液,氯化钠水溶液洗涤,有机相干燥后,pe/ea柱层析,得化合物n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ5.12(s,1h),3.70(t,j=5.7hz,2h),3.23(q,j=6.0hz,2h),1.71
–
1.66(m,2h),1.42(s,9h),0.89(s,9h),0.05(s,6h)。
[0084]
(3)称取22.0g的步骤(2)得到的n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚溶于160ml无水dmf中,冰浴下加入3.6g氢化钠(60%),搅拌下加入16.8g三氘代碘甲烷并升温到25℃反应12h,加水淬灭反应,乙酸乙酯萃取,无水硫酸钠干燥有机相,旋干溶剂得n
‑
甲基
‑
n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ3.62(t,j=6.3hz,2h),3.25(d,j=10.1hz,2h),2.85(s,3h),1.76
–
1.68(m,2h),1.44(s,9h),0.89(s,9h),0.04(s,6h)。
[0085]
(4)称取11.6g的步骤(3)得到的n
‑
甲基
‑
n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚溶于200ml四氢呋喃中,冰浴下加入44ml 1m的四丁基氟化铵四氢呋喃溶液,搅拌12h,乙酸乙酯萃取,硫酸钠干燥,旋干溶剂得n
‑
甲基
‑
n
‑
(叔丁氧羰基)乙醇胺粗品直接用于下一步反应。
[0086]
反应瓶中入7.0g n
‑
甲基
‑
n
‑
(叔丁氧羰基)丙醇胺粗品,26.1g三苯基磷,6.7g咪唑,氮气保护后,冰浴下,加入150ml溶剂二氯甲烷,搅拌,随后分批加入25.8g的i2,25℃下反应12h。之后加入纯水,终止反应,二氯甲烷萃取干燥后,pe/ea柱层析,得n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ3.31
–
3.27(m,2h),3.15(t,j=6.9hz,2h),2.87(s,3h),2.05(d,j=10.8hz,2h),1.46(s,9h)。
[0087]
制备例6 n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷的合成
[0088]
n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷结构如下所示:
[0089][0090]
制备方法如下:
[0091]
(1)称取22.0g的制备例5中步骤(2)得到的n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚溶于160ml无水dmf中,冰浴下加入3.6g氢化钠(60%),搅拌下加入16.8g三氘代碘甲烷并升温到25℃反应12h,加水淬灭反应,乙酸乙酯萃取,干燥,旋干溶剂得n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ3.62(t,j=6.3hz,2h),3.25(d,j=10.1hz,2h),1.76
–
1.68(m,2h),1.44(s,9h),0.89(s,9h),0.04(s,6h)。
[0092]
(2)称取11.6g的步骤(1)得到的n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)丙醇胺叔丁基二甲基硅醚于200ml四氢呋喃中,冰浴下加入44ml 1m的四丁基氟化铵四氢呋喃溶液,搅拌12h,乙酸乙酯萃取,硫酸钠干燥,旋干溶剂得n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)丙醇胺。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ3.84(s,1h),3.68
–
3.44(m,2h),3.41
–
3.29(m,2h),1.78
–
1.58(m,2h),1.50
–
1.36(s,9h)。
[0093]
(3)反应瓶中入7.0g步骤(2)得到的n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)丙醇胺、26g三苯基磷和6.7g咪唑,氮气保护后,冰浴下,缓慢加入150ml溶剂二氯甲烷,匀速搅拌,随后分批加入26g的i2,25℃下反应12h。之后加入纯水,终止反应,二氯甲烷萃取干燥后,pe/ea柱层析,得n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷。结构表征如下:1h nmr(600mhz,
chloroform
‑
d)δ3.31
–
3.27(m,2h),3.15(t,j=6.9hz,2h),2.05(d,j=10.8hz,2h),1.46(s,9h)。
[0094]
制备例7化合物[d6]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d6]
‑
bdmapb)的合成
[0095]
化合物[d6]
‑
bdmapb的结构如下所示:
[0096][0097]
制备方法如下:
[0098]
(1)称取5g制备例5得到的n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷和6.22g锌粉,置于50ml反应瓶中,加入10mg碘,氮气保护后,加入30ml溶剂dmf,60℃下搅拌1h。之后在25℃下静置沉淀,取上清液,加入氮气保护下,称取1.95g对溴苯偶酰、171mg醋酸钯、277mg三苯基磷和10ml dmf的反应瓶中,65℃下搅拌9h。之后利用ea
‑
h2o萃取反应液,有机相利用无水硫酸钠干燥后旋干,柱层析纯化,得化合物4,4
′‑
二(n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基乙基)苯偶酰。结构表征如下:1h nmr(600mhz,chloroform
‑
d)δ7.89(d,j=7.9hz,4h),7.36
–
7.30(m,4h),3.25(m,4h),2.84(d,j=12.9hz,6h),2.72
–
2.64(t,j=6hz,4h),1.91
–
1.82(m,4h),1.44(s,18h)。
[0099]
(2)称取500mg步骤(1)得到的4,4
′‑
二(邻苯二甲酰亚氨基丙基)苯偶酰于50ml反应瓶中,加入10ml三氟乙酸,搅拌12h。旋干溶剂,加入乙酸乙酯,滤出固体为粗品。在反应管中,加入100mg粗品,20%的氘代甲醛2ml和1ml氘代甲酸,100℃下反应12h,旋干反应溶剂,柱层析,得到化合物[d6]
‑
bdmapb,产率95%。其中[d6]指代结构中包含六个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.88(d,j=8.3hz,4h),7.47(d,j=8.2hz,4h),3.18
–
3.12(m,4h),2.88(s,6h),2.81(t,j=7.7hz,4h),2.12
–
2.03(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.38,131.33,129.83,129.01,56.97,42.04,32.05,25.40。
[0100]
制备例8化合物[d2]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d2]
‑
bdmapb)的合成
[0101]
化合物[d2]
‑
bdmapb的结构如下所示:
[0102][0103]
制备方法中除将氘代甲醛替换为等量的甲醛外,其余与制备例7一致。其中[d2]指代结构中包含两个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.88(d,j=8.3hz,4h),7.47(d,j=8.2hz,4h),3.18
–
3.12(m,4h),2.88(s,10h),2.81(t,j=7.7hz,4h),2.12
–
2.03(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.38,131.33,129.83,129.01,56.97,42.04,32.05,25.40。
[0104]
制备例9化合物[d4]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d4]
‑
bdmapb)的合成
[0105]
化合物[d4]
‑
bdmapb的结构如下所示:
[0106]
[0107]
制备方法中除将氘代甲酸替换成等量的甲酸外其余与制备例7一致。其中[d4]指代结构中包含四个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.88(d,j=8.3hz,4h),7.47(d,j=8.2hz,4h),3.18
–
3.12(m,4h),2.88(s,8h),2.81(t,j=7.7hz,4h),2.12
–
2.03(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.38,131.33,129.83,129.01,56.97,42.04,32.05,25.40。
[0108]
制备例10化合物[d
10
]
‑
4,4
′‑
双[3
‑
(二甲氨基)丙基]苯偶酰([d
10
]
‑
bdmapb)的合成
[0109]
化合物[d
10
]
‑
bdmapb的结构如下所示:
[0110][0111]
制备方法中除将制备例5得到的n
‑
甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷替换成等量的制备例6得到的n
‑
三氘代甲基
‑
n
‑
(叔丁氧羰基)氨基碘丙烷外,其余与制备例9一致。其中[d
10
]指代结构中包含十个氘原子。结构表征如下:1h nmr(600mhz,methanol
‑
d4)δ7.88(d,j=8.3hz,4h),7.47(d,j=8.2hz,4h),3.18
–
3.12(m,4h),2.88(s,2h),2.81(t,j=7.7hz,4h),2.12
–
2.03(m,4h).
13
c nmr(151mhz,methanol
‑
d4)δ194.40,148.38,131.33,129.83,129.01,56.97,42.04,32.05,25.40。
[0112]
标记效果测试:
[0113]
配制胍类化合物(胍、胍乙酸、肌酸、胍基丁酸、精氨酸、高精氨酸的浓度各为25μmol/l)的混合标准水溶液,并分别取25μl混合标准水溶液分别加入50μl的上述[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb的乙腈溶液(200μmol/l),以及25μl的naoh水溶液(1.2mol/l),在60℃下反应4小时。之后冷却至25℃,加入20μl的hcl水溶液(2mol/l),调节体系ph至4。之后将上述溶液用氮气吹干,加入等体积甲醇复溶,去除反应体系中的盐分,离心取上清液进行液相色谱
‑
四极杆飞行时间质谱联用(lc
‑
qtof ms)分析。分析条件:液相色谱仪agilent 1290、色谱柱shim
‑
pack scepter c18
‑
120(1.9μm,2.1
×
100mm)和质谱仪agilent 6545q
‑
tof ms。喷雾气压30psi,雾化气温度325℃,电压4000v。流动相a相:水(添加1%质量分数甲酸和20mm甲酸铵);b相:乙腈。洗脱梯度:0
‑
2min,b相体积分数5%;2
‑
8min,b相体积分数匀速变化至20%;8
‑
10min,b相体积分数匀速变化至100%。流速:0.4ml/min,进样量1μl。六种胍类化合物(胍、胍乙酸、肌酸、胍基丁酸、精氨酸、高精氨酸)混标与[d0]
‑
bdmapb、[d4]
‑
bdmapb、[d8]
‑
bdmapb和[d
12
]
‑
bdmapb反应得到的衍生化产物的质谱检测结果如图1
‑
6所示。从图中可以看出六种胍类化合物的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物呈现等比例的质谱峰,在m/z上呈现依次增加两个m/z单位的一组双电荷离子质谱峰,且背景噪音较低,不会对产物所在的分子量区域造成干扰。与现有衍生化试剂相比,bdmapb衍生化产物质谱响应信号增强。由此说明,本发明提供的苯偶酰类化合物能够有效的提高胍类化合物的质谱响应,有利于低峰度胍类化合物的分析检测。反应前的操作和反应后利用甲醇复溶,不仅可以有效避免蛋白质对检测的影响,也降低了检测液体系中的含盐量,避免大量的盐影响色谱柱和质谱检测。
[0114]
胍乙酸、肌酸的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物的lc结果和二级质谱结果见图7
‑
10。从图中可以看出,同一胍类化合物的不同[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物在lc中具有相似的保留行为。所有胍类化合物衍生化产物的二级质谱中,以[d0]
‑
bdmapb衍生的胍类化合物产物[m+2h]
2+
为母离子,产物二级碎裂后的特征离子为m/z 91、154、189、262和307;以[d4]
‑
bdmapb衍生的胍类化合物产物[m+2h]
2+
为母离子,二级碎裂后的特征离子为m/z 91、155、191、262和309;以[d8]
‑
bdmapb衍生的胍类化合物产物[m+2h]
2+
为母离子,二级碎裂后的特征离子为m/z 91、156、193、262和311;以[d
12
]
‑
bdmapb衍生的胍类化合物产物[m+2h]
2+
为母离子,二级碎裂后的特征离子为m/z 91、157、195、262和313。从以上结果看出,将lc行为和一级二级ms行为作为依据,可定性分析样本中所有的胍类化合物。
[0115]
线性关系考察:
[0116]
配制含六种胍类化合物(胍、胍乙酸、肌酸、胍基丁酸、精氨酸、高精氨酸)的系列浓度的混合标准溶液,同一混合标准溶液中六种胍类化合物浓度均相同,且系列浓度分别各为25、50、100、300、500、700和1000nmol/l,分别与200μmol/l[d0]
‑
bdmapb/200μmol/l[d4]
‑
bdmapb/200μmol/l[d8]
‑
bdmapb溶液混合按上述反应流程反应,200μmol/l[d
12
]
‑
bdmapb溶液与六种胍类化合物浓度各为500nmol/l的混合标准溶液反应作为内标,之后将同一化合物的所有不同反应液等体积混合氮气吹干,再用等体积甲醇复溶后进行lc
‑
qtof ms分析,分析流程与上述标记效果测试中的流程一致。以系列浓度衍生的[m+2h]
2+
离子峰高与内标的[m+2h]
2+
离子峰高之比为y轴,浓度为x轴作线性回归,结果如下:
[0117][0118][0119]
[0120][0121][0122][0123]
从以上数据可以看出上述各化合物的线性曲线的r2均达到0.99以上,显示了较强的相关性。体现了本发明提供的苯偶酰类化合物能够满足对胍类化合物的定量检测要求。
[0124]
稳定性考察:
[0125]
将上述标记效果测试中的六种胍类化合物的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生化产物待测溶液保存在4℃冰箱中,分别与24h和48h考察产物lc
‑
ms检测(检测方法与标记效果测试中相同)的稳定性,每次测试时冻存的衍生化产物待测溶液各取20μl,并加入20μl新制作的内标待测溶液,混合均匀后进样。[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb衍生化产物的内标为[d
12
]
‑
bdmapb衍生化产物,[d
12
]
‑
bdmapb衍生化产物的内标为[d0]
‑
bdmapb衍生化产物。最后将待测物峰高与内标峰高比值为纵坐标,测试时间为横坐标,制作稳定性曲线。结果如图11
‑
16。从图中可以看出,经48h后各化合物的衍生化产物的质谱信号持平,表示胍类化合物通过衍生后产物lc
‑
ms检测的稳定性高,有利于衍生化产物样品的保存和长期检测分析,说明本发明提供的苯偶酰类化合物用于胍类化合物检测的稳定性好。
[0126]
不同衍生化试剂效果比较:
[0127]
配置浓度为200μmol/l的不同衍生化试剂(苯偶酰、对甲基苯偶酰、对甲氧基苯偶酰、对氟苯偶酰、对二甲氨基苯偶酰和[d0]
‑
bdmapb),以标记效果测试中的测试方法参数对六种胍类化合物(胍、胍乙酸、肌酸、胍基丁酸、精氨酸、高精氨酸)进行检测对比产物质谱峰的信噪比。结果如下:
[0128][0129]
从表中可以看出bdmapb检测信号显著高于其他标记试剂,说明本发明提供的苯偶酰类化合物具有更好的检测灵敏度。
[0130]
小鼠血清中内源性胍类化合物的筛查:
[0131]
对4只体重在230
‑
260g之间的sd雄性大鼠腹主动脉取血,离心取上层血清,分装冻存备用。体积比以1:4的比例在血清中加入乙腈混匀,
‑
20℃冰箱过夜,沉淀蛋白,离心取上清液25μl,氮气吹干,等体积水复溶,加入50μl的[d0]
‑
bdmapb乙腈溶液(200μmol/l),以及25μl的naoh水溶液(1.2mol/l),在60℃下反应4小时。反应体系冷却至25℃,加入20μl的hcl水溶液(2mol/l),调节ph至4。上述溶液用氮气吹干,加入等体积甲醇复溶,去除反应体系中大量的盐分,离心,取上清液进行lc
‑
qtof ms检测分析(检测方法及参数与标记效果测试中的检测一致)。提取检测结果中双电荷离子,由于衍生化产物中引入相同c
24
h
32
n2o基团,不饱和度≥10,根据这两个条件对其元素组成进行初步筛查。再对满足上述条件的[m+2h]
2+
母离子进行二级碎裂,二级谱图中含有m/z 91、154、189、262、307特征碎片离子的化合物判断为胍类化合物。结果如下:
[0132][0133]
从表中可知本发明提供的苯偶酰类化合物应用在胍类化合物的检测中能够有效检测小鼠血清中的胍类化合物及其种类,显示了优秀的效果。
[0134]
肺癌患者癌症组织中六种胍类化合物的绝对定量分析:
[0135]
取三位肺癌患者的癌症组织样本,对其中六种胍类化合物进行定量。患者基本信息如下:
[0136][0137]
分析步骤如下:
[0138]
将组织样品用冰水清洗表面血水,滤纸吸去多余水分。
‑
80℃冰箱冰冻3h,冻干机冻干48h。将冻干的组织称重,每毫克组织样品中加入20μl的纯水,匀浆。匀浆后,离心取上清液,冻存备用。匀浆液以1:4的比例加入4倍体积的乙腈混匀,
‑
20℃下过夜。沉淀蛋白后离心取上清液25μl,氮气吹干,等体积水复溶,不同病人分别加入50μl的[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb乙腈溶液(200μmol/l),以及25μl的naoh水溶液(1.2mol/l),在60℃下反应4小时。配置浓度为500nmol/l的胍类化合物混标溶液,取25μl,加入50μl的[d
12
]
‑
bdmapb乙腈溶液(200μmol/l),以及25μl的naoh水溶液(1.2mol/l),在60℃下反应4h作为定量分析的内标。反应体系冷却至25℃,加入20μl的hcl水溶液(2mol/l),调节ph至4。将[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb/[d
12
]
‑
bdmapb衍生的反应液等量混匀,氮气吹干,加入与混合溶液等体积甲醇复溶,去除反应体系中大量的盐分,离心取上清液进行lc
‑
qtof ms分析。(检测方法及参数与标记效果测试中的检测一致)以[d
12
]
‑
bdmapb衍生化产物为内标,
[d0]
‑
bdmapb/[d4]
‑
bdmapb/[d8]
‑
bdmapb衍生化产物峰高与内标峰高的比值,带入线性关系考察中的线性曲线中,计算浓度,结果如下:
[0139][0140]
从表中可知本发明提供的苯偶酰类化合物应用在癌症患者中胍类化合物的检测中能够有效检测癌症组织中的胍类化合物含量,显示了优秀的应用前景。
[0141]
申请人声明,本发明通过上述实施例来说明本发明的苯偶酰类化合物及其制备方法和应用,但本发明并不局限于上述实施例,即不意味着本发明必须依赖上述实施例才能实施。所属技术领域的技术人员应该明了,对本发明的任何改进,对本发明产品各原料的等效替换及辅助成分的添加、具体方式的选择等,均落在本发明的保护范围和公开范围之内。
[0142]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
[0143]
另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1