一种丙二酸二硫酯类化合物,其制备方法及用途与流程
1.本发明涉及化学合成领域,具体涉及一种丙二酸二硫酯类化合物,其制备方法及用途。
背景技术:
2.粮食安全是关系到经济发展、社会稳定和国家安全的全局性战略问题。近年来,以稻飞虱和二化螟为主的水稻害虫抗药性迅速上升,导致施用现有农药即便增加用药量和用药次数也难以达到理想防效。因此新型高效杀虫剂的研发具有非常重要的意义。吡啶并[1,2-α]嘧啶酮介离子类化合物是一类新型杀虫剂,其化学结构式如下式v所示。2009年杜邦公司首次公开了该类化合物的杀虫活性(wo2009099929),吡啶并[1,2-α]嘧啶酮介离子类杀虫剂主要作用于烟碱乙酰胆碱受体(nachrs),但其作用机理不同于传统的新烟碱类杀虫剂,具有优异的杀虫活性,能有效防治鳞翅目和同翅目害虫。
[0003]
三氟苯嘧啶(triflumezopyrim)和dicloromezotiaz是由美国杜邦公司开发的吡啶并[1,2-α]嘧啶酮介离子杀虫剂。三氟苯嘧啶(triflumezopyrim)的化学结构式如下式(v1)所示,可用于防治水稻稻飞虱的新型杀虫剂,目前已经上市。dicloromezotiaz的化学结构式如下式(v2)所示,对鳞翅目害虫具有很好的防治效果,很可能成为防治二化螟等鱗翅目害虫的高效药剂。
[0004]
目前,吡啶并[1,2-α]嘧啶酮介离子化合物的合成是通过活泼的α芳基取代丙二酸衍生物(vi)与氨基吡啶中间体(iv)在惰性溶剂中加热缩合实现。活泼的α芳基取代丙二酸衍生物可以是α芳基取代丙二酰氯或α芳基取代丙二酸活性酯,其合成需要先通过丙二酸酯与卤代芳烃的金属偶联反应或芳基乙酸酯的克莱森缩合反应得到α芳基取代丙二酸酯,后经水解得到α芳基取代丙二酸作为原料合成,其合成路线如下式所示:
现有的吡啶并[1,2-α]嘧啶酮介离子化合物的合成过程中,由于经过了金属催化偶联,酯水解,活性酯或酰氯的制备等繁琐的步骤,整体反应收率偏低而且反应条件苛刻,而且在水解制备α芳基取代丙二酸盐的反应步骤中,除去溶剂水的工艺难度高且费时,不利于工业化生产。
[0005]
鉴于此,特提出本发明。
技术实现要素:
[0006]
本发明的首要发明目的在于提供一种丙二酸二硫酯类化合物。
[0007]
本发明的第二发明目的在于提供该化合物的制备方法。
[0008]
本发明的第三发明目的在于提供该化合物的用途。
[0009]
本发明的第四发明目的在于提供采用该化合物制备介离子类杀虫剂的方法。
[0010]
为了实现本发明的发明目的,采用的技术方案为:本发明第一方面提出一种丙二酸二硫酯衍生物,所述丙二酸二硫酯衍生物的结构式如式i所示:其中,r1选自c
1-c6烷基、c
1-c6卤代烷基、c
3-c8环烷基、c
1-c6烷氧基、c
1-c6卤代烷氧基、c
1-c6烷硫基、c
1-c6卤代烷硫基、未取代的或被(r4)m取代的芳基、芳氧基、芳c
1-c6烷基或杂芳基;m选自1、2、3、4或5,当m>1时r4可相同或不同;r2、r3各自独立地选自c
1-c6烷基、c
1-c6卤代烷基、未取代的或被(r5)r取代的芳基、芳c
1-c6烷基或杂芳基;r选自1、2、3、4或5,当r>1时r5可相同或不同;
r4、r5各自独立地选自卤素、氰基、氨基、硝基、羟基、羧基、酯基、c
1-c6烷基、c
1-c6卤代烷基、c
3-c8环烷基、c
1-c6烷氧基、c
1-c6烷硫基、c
1-c6卤代烷氧基、c
1-c6卤代烷硫基;但,r1为甲基时,r2与r3不同时为乙基、苯基、苄基、对甲基苯基或4-氯苯基;或者,r1为乙基时,r2与r3不同时为苯基、4-溴苯基或3,4-二氯苯基;或者,r1为丙基或丁基时,r2与r3不同时为乙基;或者,r1为异丙基时,r2与r3不同时为苯基;或者,r1为苯基时,r2与r3不同时为苯基或对甲苯基。
[0011]
本发明第二方面提出该丙二酸二硫酯衍生物的制备方法,所述制备方法包括采用如式ii所示的化合物与如式iii所示的化合物发生偶联反应,制备得到如式ⅰ所示的化合物,化学反应方程式如式a所示:其中,lg表示离去基团;所述离去基团选自对硝基苯酚基、卤素、酰氧基、硫代酰氧基、对甲苯磺酰氧基、咪唑基、吡啶基或嘧啶基。
[0012]
本发明第三方面提出该丙二酸二硫酯衍生物在用于合成介离子类杀虫剂的应用。
[0013]
本发明第四方面提出一种该丙二酸二硫酯衍生物合成介离子类杀虫剂的方法,使用式ⅰ所示化合物作为中间体,与式ⅳ所示的氨基吡啶中间体进行缩合反应,合成如式
ⅴ
所示的吡啶并[1,2-α]嘧啶酮介离子类化合物,化学反应方程式如式b所示:。
[0014]
本发明至少具有以下有益的效果:本发明丙二酸二硫酯类化合物比丙二酰氯更加稳定,且比普通丙二酸酯衍生物反应活性更高的优势,可用作合成吡啶并[1,2-α]嘧啶酮介离子型化合物的优良中间体。
[0015]
本发明的丙二酸二硫酯类化合物的合成方法以简单和低成本的化合物作为起始原料,可一步高收率得到的本发明的丙二酸二硫酯化合物,采用本发明的丙二酸二硫酯类化合物制备吡啶并[1,2-α]嘧啶酮介离子型化合物,相较于现有方法具有步骤简单、条件温和、高收率等优势。
具体实施方式
[0016]
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。另外,为了更好的说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体
细节,本发明同样可以实施。在一些实施例中,对于本领域技术人员熟知的原料、元件、方法、手段等未作详细描述,以便于凸显本发明的主旨。
[0017]
本发明实施例提出一种丙二酸二硫酯衍生物,可以用于更加简单、条件温和、高效合成吡啶并[1,2-α]嘧啶酮介离子化合物,其结构式如式i所示:其中,r1选自c
1-c6烷基、c
1-c6卤代烷基、c
3-c8环烷基、c
1-c6烷氧基、c
1-c6卤代烷氧基、c
1-c6烷硫基、c
1-c6卤代烷硫基、未取代的或被(r4)m取代的芳基、芳氧基、芳c
1-c6烷基或杂芳基;m选自1、2、3、4或5,当m>1时r4可相同或不同;r2、r3各自独立地选自c
1-c6烷基、c
1-c6卤代烷基、未取代的或被(r5)r取代的芳基、芳c
1-c6烷基或杂芳基;r选自1、2、3、4或5,当r>1时r5可相同或不同;r4、r5各自独立地选自卤素、氰基、氨基、硝基、羟基、羧基、酯基、c
1-c6烷基、c
1-c6卤代烷基、c
3-c8环烷基、c
1-c6烷氧基、c
1-c6烷硫基、c
1-c6卤代烷氧基、c
1-c6卤代烷硫基;但,r1为甲基时,r2与r3不同时为乙基、苯基、苄基、对甲基苯基或4-氯苯基;或者,r1为乙基时,r2与r3不同时为苯基、4-溴苯基或3,4-二氯苯基;或者,r1为丙基或丁基时,r2与r3不同时为乙基;或者,r1为异丙基时,r2与r3不同时为苯基;或者,r1为苯基时,r2与r3不同时为苯基或对甲苯基。
[0018]
其中,c
1-c6烷基可选自甲基、乙基、丙基、异丙基、丁基、叔丁基、戊基、己基,c
1-c6卤代烷基可选自一氟甲基、二氟甲基、三氟甲基、二氟甲基、三氟乙基、七氟异丙基,c
3-c8环烷基可选自环丙基、环丁基、环戊基、环己基,芳基可选自苯基、萘基,芳甲基可选自苯甲基、萘甲基,c
1-c6烷氧基可选自甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、叔丁氧基、戊氧基、己氧基,c
1-c6烷硫基可选自甲硫基、乙硫基、丙硫基、异丙硫基、丁硫基、叔丁硫基、戊硫基、己硫基,c
1-c6卤代烷硫基可选自二氟甲硫基、二氟乙硫基、三氟甲硫基、三氟乙硫基、三氟丙硫基、七氟异丙基,c
6-c
10
芳氧基可选自苯氧基、萘氧基,c
3-c
10
芳杂基中的杂原子可选自氮原子、硫原子、氧原子,c
3-c
10
芳杂基可选自呋喃、噻吩、吡咯、噻唑、咪唑、吡啶、吡嗪、嘧啶、哒嗪、吲哚、喹啉、蝶啶、吖啶等,芳c
1-c6烷基可选自苯甲基、苯乙基、苯丙基、苯异丙基、苯丁基、苯叔丁基、苯戊基、苯己基;卤素选自氟、氯、溴、碘。
[0019]
优选的,r1选自取代或未取代的苯基、取代或未取代的萘基、取代或未取代的c
1-c4烷基、取代或未取代的苯氧基。
[0020]
具体的,丙二酸二硫酯衍生物的结构式如式ia所示:
ꢀꢀ
ꢀ
还可以选择如表1所示的化合物:表1
ꢀ
ꢀꢀꢀ
ꢀꢀꢀ
ꢀꢀ
ꢀ
ꢀꢀꢀ
ꢀꢀꢀ
ꢀ
ꢀ
ꢀ
丙二酸二硫酯衍生物还可选自如式ib、式ic、式id、式ie、式if、式ig或式ih所示的化合物:其中,r2、r3各自独立地选自甲基、乙基、正丙基、异丙基、正丁基、异丁基、叔丁基、
苯基、苯甲基、对甲苯基、对甲氧基苯基、对氯苯基、三氟乙基或2-吡啶基;优选乙基或对甲苯基。
[0022]
可选自以下结构式所示的化合物:其中优选化合物如表2所示:表2还可以选自表3所示的化合物:
本发明实施例还涉及该丙二酸二硫酯衍生物的制备方法,制备方法包括采用如式ii所示的化合物与如式iii所示的化合物发生偶连反应,制备得到如式ⅰ所示的化合物,化学反应方程式如式a所示:其中,lg表示离去基团;r1、r2、r3具有式ⅰ中相同的含义。
[0023]
离去基团选自对硝基苯酚基(-oc6h4no2)、卤素(-x)、酰氧基(-ocor)、硫代酰氧基(-scor)、对甲苯磺酰氧基(-ots)、咪唑基(-c3h3n2)、吡啶基(-c5h4n)、嘧啶基(-c4h3n2)等。
[0024]
偶联反应的反应条件包括在条件i或条件ii下进行:条件i:式ⅱ所示化合物作为酰化试剂与式ⅲ进行直接偶联得到式ⅰ所示化合物,其中涉及在弱碱作用下,路易斯酸参与式ⅲ所示化合物的烯醇化,反应过程如下。
78℃~ 300℃,优选0℃~ 200℃,更优选有机溶剂的回流温度;反应结束后冷却至室温,加入石油醚析出固体,过滤、干燥,即得吡啶并[1,2-α]嘧啶酮介离子类化合物。
[0037]
本发明实施例以简单和低成本的式ⅱ、式ⅲ化合物为起始原料,一步高收率得到的式ⅰ丙二酸二硫酯化合物是新化合物;式ⅰ丙二酸二硫酯化合物具有比丙二酰氯更加稳定,且比普通丙二酸酯衍生物反应活性更高的优势,可用作合成吡啶并[1,2-α]嘧啶酮介离子型化合物的优良中间体。相较于现有方法具有步骤简单、条件温和、高收率等优势。
[0038]
下文的实施例阐述本发明但不以任何方式限制本发明。
[0039]
为恰当地验证该反应路线,将合成中间体及部分介离子型吡啶并[1,2-α]嘧啶酮化合物进行分离并表征。通过常规波谱技术确认所述化合物的结构:质子nmr(s=单峰;d=双峰;dd=双双峰;t=三重峰;q=四重锋);碳nmr(s=单峰;d=双峰;dd=双双峰;t=三重峰;q=四重锋)。
[0040]
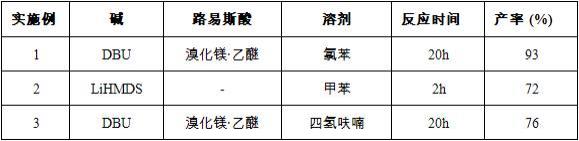
实施例1 2-苯基丙二酸二乙硫酯的制备1向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到50 mg黄色油状物,收率93%。1h nmr (400 mhz, chloroform-d) δ 7.5
ꢀ–ꢀ
7.4 (m, 2h), 7.4 (dd, j = 5.11, 2.07 hz, 3h), 4.9 (s, 1h), 2.9 (qd, j = 7.45, 1.36 hz, 4h), 1.2 (t, j = 7.46 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.0, 132.2, 129.4, 128.7, 128.7, 72.6, 24.3, 14.2 ppm ; esi-ms, m/z:291.03[m+na]
+ ; ir (kbr): v = 3062, 2969, 2930, 2874, 1700, 1600, 1452, 1377, 869, 725 cm-1
; rf = 0.52(石油醚:乙酸乙酯=10:1)。
[0041]
实施例2 2-苯基丙二酸二乙硫酯的制备2向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、甲苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、双(三甲基硅基)胺基锂(lihmds)(0.3mmol),室温搅拌2 h。加入饱和氯化铵溶液猝灭反应,用乙酸乙酯萃取2次,加入饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到39 mg黄色油状物,收率72%。其表征数据如实施例1。
[0042]
实施例3 2-苯基丙二酸二乙硫酯的制备3
向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、四氢呋喃1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到41 mg黄色油状物,收率76%。其表征数据如实施例1。
[0043]
实施例4 2-苯基丙二酸二乙硫酯的制备4向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和n,n-二甲基乙醇胺(dabco)(67 mg, 0.6mmol),室温搅拌20 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到13 mg黄色油状物,收率24%。其表征数据如实施例1。
[0044]
实施例5 2-苯基丙二酸二乙硫酯的制备5向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二异丙基乙胺(dipea)(77.4 mg, 0.6mmol),室温搅拌20.5 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯= 20:1)分离得到40.2 mg黄色油状物,收率75%。其表征数据如实施例1。
[0045]
实施例6 2-苯基丙二酸二乙硫酯的制备6
向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、4-二甲基氨基吡啶(dmap)(7mg, 0.6mmol)和二异丙基乙胺(dipea)(77.4 mg, 0.6mmol),室温搅拌22 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到35.5 mg黄色油状物,收率66%。其表征数据如实施例1。
[0046]
实施例7 2-苯基丙二酸二乙硫酯的制备7向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、和三乙胺(dipea)(60.6 mg, 0.6mmol),室温搅拌22 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到36 mg黄色油状物,收率67%。其表征数据如实施例1。
[0047]
实施例8 2-苯基丙二酸二乙硫酯的制备8向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌24 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到40.2 mg黄色油状物,收率80%。其表征数据如实施例1。
[0048]
实施例9 2-苯基丙二酸二乙硫酯的制备9
向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、甲苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌19 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到20.3 mg黄色油状物,收率38%。其表征数据如实施例1。
[0049]
实施例10 2-苯基丙二酸二乙硫酯的制备10向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、四氢呋喃1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到40.9 mg黄色油状物,收率76%。其表征数据如实施例1。
[0050]
实施例11 2-苯基丙二酸二乙硫酯的制备11向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、乙腈1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)、和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20.5 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到44.5 mg黄色油状物,收率83%。其表征数据如实施例1。
[0051]
实施例12 2-苯基丙二酸二乙硫酯的制备12
向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(45 mg, 0.3 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20.5 h。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到45.9 mg黄色油状物,收率86%。
[0052]
实施例13 2-苯基丙二酸二乙硫酯的制备13向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、四氢呋喃2 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、双(三甲基硅基)胺基钠(nahmds)(0.3mmol),室温搅拌3 h。加入饱和氯化铵溶液猝灭反应,用乙酸乙酯萃取2次,加入饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到6 mg黄色油状物,收率11%。其表征数据如实施例1。
[0053]
实施例14 2-苯基丙二酸二乙硫酯的制备14向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、二氯甲烷1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、双(三甲基硅基)胺基锂(lihmds)(0.3mmol),室温搅拌0.5 h。加入饱和氯化铵溶液猝灭反应,用乙酸乙酯萃取2次,加入饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到29 mg黄色油状物,收率54%。其表征数据如实施例1。
[0054]
表4为实施例1-14的反应条件及产率汇总表表4
hz, 4h), 1.26 (t, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 192.7, 132.8, 128.6, 127.1, 126.8, 67.3, 24.5, 14.2 ppm ; esi-ms,m/z:296.96[m+na]
+
; ir (kbr): v = 3071, 2931, 2875, 1699, 1664, 1613, 1450, 1376, 897, 760, 687, 522 cm-1
; rf = 0.47 (石油醚:乙酸乙酯=10:1).实施例19 2-(1-萘基)丙二酸二乙硫酯的制备向单口瓶中依次加入1-萘乙酸乙硫酯(46 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌26 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到40 mg油状化合物, 收率63%。1h nmr (400 mhz, chloroform-d) δ 8.0
ꢀ–ꢀ
8.0 (m, 1h), 7.9
ꢀ–ꢀ
7.9 (m, 2h), 7.7 (dd, j = 7.32, 1.22 hz, 1h), 7.6
ꢀ–ꢀ
7.5 (m, 3h), 5.8 (s, 1h), 3.1
ꢀ–ꢀ
2.6 (m, 4h), 1.2 (t, j = 7.42 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.8, 134.0, 131.7, 129.5, 129.2, 128.3, 127.7, 127.0, 126.0, 125.4, 122.7, 68.3, 24.4, 14.2 ppm ; esi-ms, m/z:341.06[m+na]
+
; ir (kbr): v = 3050, 1694, 1595, 1508, 1448, 1374, 886, 796 cm-1
; rf = 0.42 (石油醚:乙酸乙酯=10:1).实施例20 2-(2-萘基)丙二酸二乙硫酯的制备向单口瓶中依次加入2-萘乙酸乙硫酯(46 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌27 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到47.8 mg淡黄色固体,收率75%。1h nmr (400 mhz, chloroform-d) δ 7.96
ꢀ–ꢀ
7.78 (m, 4h), 7.65
ꢀ–ꢀ
7.45 (m, 3h), 5.11 (s, 1h), 2.92 (q, j = 7.4 hz, 4h), 1.23 (t, j = 7.4 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.1, 133.2, 129.7, 129.0, 128.5, 128.2, 127.7, 126.7, 126.6, 126.4, 72.6, 24.4, 14.3 ppm ; esi-ms, m/z:341.05[m+na]
+
; ir (kbr): v = 2966, 2861, 1690, 1500, 1448, 1372, 797, 763 cm-1
; rf = 0.42 (石油醚:乙酸乙酯=10:1).
实施例21 2-(4-氯苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入4-氯苯乙酸乙硫酯(43 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌16 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到51 mg无色油状物,收率84%。1h nmr (400 mhz, chloroform-d) δ 7.46
ꢀ–ꢀ
7.29 (m, 4h), 4.92 (s, 1h), 2.91 (q, j = 7.4 hz, 4h), 1.24 (t, j = 7.4 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 192.5, 134.9, 130.7, 130.6, 128.9, 71.7, 24.4, 14.2 ppm ; esi-ms, m/z: 325.00 [m+na]
+
; ir (kbr): v = 2930,2873, 1665, 1452, 1376, 874, 830, 693 cm-1
; rf = 0.47 (石油醚:乙酸乙酯=10:1).实施例22 2-(4-甲基苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入4-甲基苯乙酸乙硫酯(38.8 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌16 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到45.2 mg无色油状物,收率80%。1h nmr (400 mhz, chloroform-d) δ 7.34 (d, j = 8.14 hz, 2h), 7.23
ꢀ–ꢀ
7.15 (m, 2h), 4.90 (s, 1h), 2.97
ꢀ–ꢀ
2.85 (m, 4h), 2.35 (s, 3h), 1.23 (t, j = 7.44 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.3, 138.7, 129.5, 129.3, 129.3, 72.2, 24.3, 21.2, 14.3 ppm ; esi-ms, m/z: 281.05 [m-h]-; ir (kbr): v = 2969, 2929, 2873, 1693, 1512, 1453, 1376, 873, 758, 693 cm-1
; rf = 0.57 (石油醚:乙酸乙酯=10:1).实施例23 2-(4-甲氧基苯基)丙二酸二乙硫酯的制备
向单口瓶中依次加入4-甲氧基苯乙酸乙硫酯(42 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌17 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到50.2 mg无色油状物,收率84%。1h nmr (400 mhz, chloroform-d) δ 7.37 (d, j = 8.80 hz, 2h), 6.90 (d, j = 8.76 hz, 2h), 4.88 (s, 1h), 3.81 (s, 3h), 2.90 (q, j = 7.33 hz, 4h), 1.23 (t, j = 7.46 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.6, 160.0, 130.7, 124.3, 114.1, 71.7, 55.3, 24.3, 14.3 ppm ; esi-ms, m/z: 297.06 [m-h]-; ir (kbr): v = 2967, 2836, 1663, 1608, 1452, 1376, 1303, 1249, 876, 70 cm-1
; rf = 0.36 (石油醚:乙酸乙酯=10:1).实施例24 2-(2-氟苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入2-氟苯乙酸乙硫酯(39.4 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌19 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到47.6 mg无色油状物,收率83%。1h nmr (400 mhz, chloroform-d) δ 7.37
ꢀ–ꢀ
7.29 (m, 1h), 7.22 (d, j = 1.10 hz, 2h), 7.11
ꢀ–ꢀ
7.00 (m, 1h), 4.94 (s, 1h), 2.92 (q, j = 7.44 hz, 4h), 1.25 (t, j = 7.44 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 192.3, 162.7 (d, j = 246.90 hz), 134.2 (d, j = 7.93 hz), 130.1 (d, j = 8.22 hz), 125.2 (d, j = 3.09 hz), 116.5 (d, j = 22.81 hz), 115.7 (d, j = 20.95 hz), 72.0 (d, j = 1.87 hz), 24.4, 14.2 ppm ; esi-ms, m/z: 209.02 [m+na]
+
; ir (kbr): v = 2964, 2927, 2874, 1695, 1660, 1447, 1375, 1260, 1136, 967, 871, 758, 686 cm-1
; rf = 0.50 (石油醚:乙酸乙酯=10:1).实施例25 2-(4-溴苯基)丙二酸二乙硫酯的制备
向单口瓶中依次加入4-溴苯乙酸乙硫酯(51.8 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到55.2 mg无色油状物,收率80%。1h nmr (400 mhz, chloroform-d) δ 7.50 (d, j = 8.50 hz, 2h), 7.34 (d, j = 8.50 hz, 2h), 4.90 (s, 1h), 2.91 (q, j = 7.44 hz, 4h), 1.24 (t, j = 7.44 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 192.4, 131.9, 131.1, 131.0, 123.1, 71.8, 24.4, 14.2 ppm ; esi-ms, m/z: 368.97 [m+na]
+
; ir (kbr): v = 2969, 2929, 2872, 1665, 1588, 1486, 1451, 1375, 1260, 985, 871, 768, 689 cm-1
; rf = 0.56 (石油醚:乙酸乙酯=10:1).实施例26 2-环丙基丙二酸二乙硫酯的制备向单口瓶中依次加入环丙基乙酸乙硫酯(22.8 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌17 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到17.1 mg无色油状物,收率37%。1h nmr (400 mhz, chloroform-d) δ 7.3 (s, 1h), 2.9 (q, j = 7.44 hz, 4h), 2.9 (d, j = 10.43 hz, 1h), 1.3 (t, j = 7.43 hz, 6h), 0.8
ꢀ–ꢀ
0.7 (m, 2h), 0.4
ꢀ–ꢀ
0.3 (m, 2h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 194.6, 72.5, 23.9, 14.4, 11.2, 4.8 ppm ; esi-ms, m/z: 255.02 [m+na]
+
; ir (kbr): v = 3082, 2968, 2930, 2873, 1670, 1414, 1375, 1078, 971, 830, 761, 720 cm-1
; rf = 0.48 (石油醚:乙酸乙酯=10:1).实施例27 s-乙基-s
’‑
对甲苯基-2-乙基丙二酸二硫酯的制备氩气保护下,向单口瓶中依次加入丁酸乙硫酯(26.5 mg, 0.2 mmol)、甲苯 1 ml、双甲硅基氨基锂 lihmds ( 0.3 mmol , 1m) 、乙基1h-咪唑-1-硫酯(62 mg, 0.4 mmol),
室温搅拌3 h,tlc监测反应结束。加入饱和氯化铵溶液猝灭反应,用乙酸乙酯萃取2次,加入饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =50:1)分离得到17.5 mg无色油状物, 收率40 % 。1h nmr (400 mhz, chloroform-d) δ 3.68 (t, j = 7.42 hz, 1h), 2.92 (q, j = 7.35 hz, 4h), 2.00 (p, j = 7.40 hz, 2h), 1.26 (t, j = 7.48 hz, 6h), 0.95 (t, j = 7.43 hz, 3h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 194.2, 70.0, 53.4, 23.9, 14.4, 11.7 ppm ; ir: (kbr) v= 2969, 2931,2875, 1698, 1670, 1455, 1377, 1262, 970, 874, 796 cm-1 ; esi-ms,m/z: 243.03 [m+na]
+ ; rf= 0.81 (石油醚:乙酸乙酯=10:1)。
[0058]
实施例28 2-(4-氟苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入4-氟苯乙酸乙硫酯(39.4 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到46 mg无色油状物,收率80%。1h nmr (400 mhz, chloroform-d) δ 7.44 (dd, j = 8.80, 5.25 hz, 2h), 7.06 (t, j = 8.67 hz, 2h), 4.93 (s, 1h), 2.91 (q, j = 7.41 hz, 4h), 1.24 (t, j = 7.45 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 192.9, 161.8, 131.2 (d, j = 8.21 hz), 128.0 (d, j = 3.32 hz), 115.7 (d, j = 21.69 hz), 71.6, 24.4, 14.2 ppm ; ir (kbr): v = 3075, 1691, 1602, 1540, 1452, 1377, 850, 777, 697 cm-1
; esi-ms, m/z: 285.03 [m-h]-; rf = 0.41 (石油醚:乙酸乙酯=10:1).实施例29 2-(4-氰基苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入4-氰基乙酸乙硫酯(41 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌20.5 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到46 mg无色油状物,收率
834, 724, 627 cm-1 ; rf = 0.40 (石油醚:乙酸乙酯=10:1).实施例32 s-乙基-s
’‑
丙基-2-苯基丙二硫酯的制备向单口瓶中依次加入苯乙酸乙硫酯(36mg, 0.2 mmol)、氯苯1 ml、s-丙基1h-咪唑-1-硫酯(68 mg,0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌21 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到47.8 mg无色油状化合物, 收率85%。1h nmr (400 mhz, chloroform-d) δ 7.5
ꢀ–ꢀ
7.4 (m, 2h), 7.4 (dd, j = 5.03, 1.90 hz, 3h), 4.96 (s, 1h),2.9
ꢀ–ꢀ
2.8 (m, 4h), 1.6
ꢀ–ꢀ
1.5 (m, 2h), 1.2 (t, j = 7.44 hz, 3h), 0.9 (t, j = 7.37 hz, 3h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 193.0, 193.0, 132.3, 129.4, 128.7, 128.7, 72.6, 31.7, 24.3, 22.6, 14.2, 13.3 ppm ; esi-ms, m/z: 281.06 [m-h]-; ir (kbr): v = 3061, 2967, 2851, 1702, 1665, 1493, 1454, 1376, 879, 725 cm-1 ; rf = 0.47 (石油醚:乙酸乙酯=10:1).实施例33 s-乙基-s
’‑
叔丁基-2-苯基丙二硫酯的制备向单口瓶中依次加入苯乙酸乙硫酯(36 mg, 0.2 mmol)、氯苯1 ml、s-叔丁基1h-咪唑-1-硫酯(74 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌24 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到49.5 mg无色油状化合物i17, 收率90%。1h nmr (400 mhz, chloroform-d) δ 7.4 (dd, j = 7.61, 2.19 hz, 2h), 7.4
ꢀ–ꢀ
7.3 (m, 3h), 4.9 (s, 1h), 2.9 (q, j = 7.45 hz, 2h), 1.5 (s, 9h), 1.2 (t, j = 7.44 hz, 3h) ppm ;
13
c nmr (101 mhz, chloroform-d) δ 193.0, 192.9, 132.4, 129.5, 129.4, 128.7, 128.6, 128.5, 127.2, 73.0, 51.1, 49.3,48.3; 24.3, 14.2 ppm ; esi-ms, m/z: 295.09 [m-h]-; ir (kbr): v = 3062, 1697, 1599, 1491, 1454, 1375, 869, 830, 724 cm-1 ; rf = 0.61 (石油醚:乙酸乙酯=10:1).实施例34 2-苯氧基丙二酸二乙硫酯的制备
向单口瓶中依次加入苯氧乙酸乙硫酯(39.2 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环(dbu)(90 μl, 0.6 mmol),室温搅拌17 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到40 mg无色油状化合物,收率70%。1h nmr (400 mhz, chloroform-d) δ 7.3 (dd, j = 8.74, 7.40 hz,2h), 7.1
ꢀ–ꢀ
7.0 (m,1h), 7.0 (dt, j = 7.77, 1.03 hz, 2h), 5.2 (s, 1h), 2.9 (q, j = 7.46 hz, 4h), 1.3 (t, j = 7.45 hz, 6h) ppm ; 13
c nmr (101 mhz, cdcl3) δ 193.7, 156.7, 129.9, 123.2, 115.8, 88.3, 23.3, 14.1 ppm ; esi-ms, m/z: 283.04 [m-h]-; ir (kbr): v = 2969, 2873, 1700, 1454, 1375, 1260, 874, 688 cm-1
; rf = 0.30 (石油醚:乙酸乙酯=10:1).实施例35 2-甲氧基丙二酸二乙硫酯的制备向单口瓶中依次加入甲氧基乙酸乙硫酯(50 mg, 0.37 mmol)、氯苯 2 ml、s-乙基1h-咪唑-1-硫酯(116.4 mg, 0.75 mmol)、溴化镁
·
乙醚(241 mg, 0.93 mmol)和dbu(191 μl, 1.12 mmol),室温搅拌24 h,tlc监测反应结束。加入乙酸(1.48 mmol),搅拌5分钟,加入乙酸乙酯(4 ml)和水(4ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到46 mg黄色油状化合物,收率55%。1h nmr (400 mhz, chloroform-d) δ 4.34 (s, 1h), 3.59 (s, 3h), 2.91 (qd, j = 7.37, 2.83 hz, 4h), 1.27 (t, j = 7.44 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 194.6, 92.1, 59.8, 23.0, 14.2 ppm ; ir (kbr): v = 2967, 2931, 2874, 1700, 1453, 1376, 1134, 1080, 991, 803, 634 cm-1 ; esi-ms, m/z: 221.11 [m-h]-。rf= 0.32 (石油醚:乙酸乙酯=10:1)。
[0059]
实施例36 2-(3,5-二氯苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入3,5-二氯苯乙酸乙硫酯(50 mg, 0.2 mmol)、氯苯1 ml、s-乙基1h-咪唑-1-硫酯(60 mg, 0.4 mmol)、溴化镁
·
乙醚(129 mg, 0.5 mmol)和二氮杂二环
(dbu)(90 μl, 0.6mmol),室温搅拌2 0 h,tlc监测反应结束。加入乙酸(0.8 mmol),搅拌5分钟,加入乙酸乙酯(2 ml)和水(2 ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到46 mg黄色油状化合物,收率68%。1h nmr (400 mhz, chloroform-d) δ 7.37 (d, j = 1.90 hz, 2h), 7.35 (t, j = 1.86 hz, 1h), 4.87 (s, 1h), 2.93 (dd, j = 7.41, 1.02 hz, 4h), 1.26 (t, j = 7.47 hz, 6h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 191.4, 135.1, 135.0, 128.9, 127.8, 71.4, 24.5, 14.1 ppm ; esi-ms, m/z: 335.08 [m-h]
‑ꢀ
; ir (kbr): v = 3078, 1698, 1453, 1377, 860, 796, 695, 580 cm-1
; rf = 0.44 (石油醚:乙酸乙酯=10:1).实施例37 2-(3,5-二氯苯基)丙二酸二乙硫酯的制备向单口瓶中依次加入3,5-二氯苯乙酸乙硫酯(100 mg, 0.4 mmol)、干燥的四氢呋喃2 ml、s-乙基1h-咪唑-1-硫酯(72 mg, 0.48 mmol)、双(三甲基硅基)胺基锂(lihmds)(0.48 mmol),室温反应3 h,tlc监测反应结束。加入饱和氯化铵溶液猝灭反应,用乙酸乙酯萃取2次,加入饱和食盐水洗涤,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到80 mg黄色油状物, 收率59%,其表征数据如实施例36。
[0060]
实施例38 s-乙基-s
’‑
对甲苯基-2-甲基丙二酸二硫酯的制备向单口瓶中依次加入乙酸对甲苯硫酯(70 mg, 0.39 mmol)、二氯甲烷 2 ml、s-乙基1h-咪唑-1-硫酯(116.5 mg, 0.78 mmol)、溴化镁
·
乙醚(260 mg, 0.97 mmol)和三乙胺(162 μl, 1.17mmol),室温搅拌25 h,tlc监测反应结束。加入乙酸(1.56 mmol),搅拌5分钟,加入乙酸乙酯(4 ml)和水(4ml)分液萃取2次,有机相用无水硫酸钠干燥,减压蒸除溶剂。残余物经硅胶柱色谱(石油醚:乙酸乙酯 =20:1)分离得到33 mg无色油状化合物,收率32%。1h nmr (400 mhz, chloroform-d) δ 7.3 (d, j = 8.19 hz, 2h), 7.2
ꢀ–ꢀ
7.2 (m,2h), 3.9 (q, j = 7.06 hz, 1h), 2.9 (q, j = 7.42 hz, 2h), 1.5 (d, j = 7.03 hz, 3h), 1.3 (t, j = 7.43 hz, 3h) ppm ;
13
c nmr (101 mhz, chloroform-d) δ 194.8, 193.6, 140.0, 134.4, 130.1, 123.4, 61.7, 23.9, 21.3, 14.7, 14.4 ppm ; esi-ms, m/z: 291.06 [m+na]
+
; rf = 0.50 (石油醚:乙酸乙酯=10:1).实施例39 介离子杀虫剂三氟苯嘧啶(triflumezopyrim)v1的制备1
向10 ml反应瓶中依次加入2-(3-三氟甲基)苯基丙二酸二乙硫酯(76 mg, 0.22 mmol)、1 ml氯苯和n-(嘧啶-5-基甲基)吡啶-2-胺(21 mg, 0.11 mmol),回流搅拌3小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得39 mg黄色晶状固体triflumezopyrim (收率87%)。 1
h nmr (400 mhz, dmso-d6) δ 9.35 (dd, j = 6.89, 1.63 hz, 1h), 9.11 (s, 1h), 8.83 (s, 2h), 8.30 (ddd, j = 8.89, 7.05, 1.69 hz, 1h), 8.19 (d, j = 1.81 hz, 1h), 8.13 (d, j = 7.74 hz, 1h), 7.88 (d, j = 8.96 hz, 1h), 7.71
ꢀ–ꢀ
7.38 (m, 3h), 5.62 (s, 2h) ppm ; 13
c nmr (151 mhz, dmso-d6) δδ 159.1, 157.9, 156.3, 154.2, 146.9, 144.7, 137.1, 134.7, 132.0, 130.3, 128.4, 128.3 (q, j = 31.01hz), 127.1 (q, j = 4.06 hz), 125.1 (q, j=270 hz), 122.1 (q, j = 3.66 hz), 117.2, 114.7, 92.6, 42.1, 40.5 ppm .实施例40 介离子杀虫剂三氟苯嘧啶(triflumezopyrim)v1的制备2向10 ml反应瓶中依次加入s-乙基-s
’‑
对甲苯基-2-(3-三氟甲基)丙二酸二硫酯(40 mg, 0.12 mmol)、1 ml甲苯和n-(嘧啶-5-基甲基)吡啶-2-胺(19 mg, 0.10 mmol),回流搅拌24小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得27 mg黄色晶状固体triflumezopyrim(收率68%)。其表征如实施例39。
[0061]
实施例41 介离子杀虫剂三氟苯嘧啶(triflumezopyrim)v1的制备3 (克级反应)向100 ml反应瓶中依次加入2-(3-三氟甲基)苯基丙二酸二乙硫酯(2.837 g, 8.44 mmol)、30 ml氯苯和n-(嘧啶-5-基甲基)吡啶-2-胺(0.784 g, 4.215 mmol),回流搅拌10小时,反应液冷却至室温,加入石油醚析出固体,过滤、干燥得到1.414 g 黄色晶状固体triflumezopyrim(收率84%),其表征数据如实施例39。
[0062]
与现有技术比较
*数据摘录自pest manag sci2017, 73, 796
–
806.实施例42 介离子杀虫剂dicloromezotiaz (v2)的制备1向10 ml反应瓶中依次加入2-(3,5-二氯苯基)丙二酸二乙硫酯(58 mg, 0.17 mmol)、1 ml 1,3,5-三甲苯和n-(((2-氯噻唑-5-基)甲基)-3-甲基吡啶-2-胺(21 mg, 0.09 mmol),回流搅拌6小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得24 mg黄色晶状固体dicloromezotiaz(收率62%)。1h nmr (600 mhz, chloroform-d) δ 9.75
ꢀ–ꢀ
9.05 (m, 1h), 8.02 (d, j = 7.20 hz, 1h), 7.77 (d, j = 1.95 hz, 2h), 7.45 (s, 1h), 7.40 (t, j = 6.96 hz, 1h), 7.23 (t, j = 1.93 hz, 1h), 5.59 (s, 2h), 2.82 (s, 3h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 160.7, 154.1, 153.2, 148.3, 147.3, 140.0, 137.0, 136.4, 134.0, 131.8, 128.8, 126.2, 124.0, 116.8, 92.4, 44.3, 23.6 ppm.实施例43介离子杀虫剂dicloromezotiaz (v2)的制备2向10 ml反应瓶中依次加入2-(3,5-二氯苯基)丙二酸二乙硫酯(85 mg, 0.25 mmol)、1 ml 氯苯和n-(((2-氯噻唑-5-基)甲基)-3-甲基吡啶-2-胺(30 mg, 0.13 mmol),回流搅拌24小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得27 mg黄色晶状固体dicloromezotiaz(收率47%)。其表征数据如实施例42。
[0063]
实施44 介离子杀虫剂dicloromezotiaz (v2)的制备3向10 ml反应瓶中依次加入s-乙基-s
’‑
对甲苯基-2-(3,5二氯苯基)丙二酸二硫酯(40 mg, 0.10 mmol)、2 ml 氯苯和n-(((2-氯噻唑-5-基)甲基)-3-甲基吡啶-2-胺(24 mg, 0.10 mmol),回流搅拌8小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得12 mg黄色晶状固体dicloromezotiaz(收率27%)。其表征数据如实施例42。
[0064]
与现有技术比较
*数据摘录自pest manag sci2017, 73, 796
–
806.实施例45介离子杀虫剂v3的制备向10 ml反应瓶中依次加入2-苯基丙二酸二乙硫酯(73 mg, 0.27 mmol)、2 ml 氯苯和n-苄基吡啶-2-胺(25 mg, 0.14 mmol),回流搅拌3小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得42 mg黄色晶状固体1-苄基-4-氧代-3-苯基-4h-吡啶并[1,2-α]嘧啶-1-鎓-2-盐(v3)(收率93%),黄色晶状固体。1h nmr (400 mhz, chloroform-d) δ 9.52 (ddd, j = 6.9, 1.7, 0.6 hz, 1h), 7.93 (ddd, j = 8.8, 7.0, 1.7 hz, 1h), 7.82 (dd, j = 8.3, 1.3 hz, 2h), 7.44
ꢀ–ꢀ
7.26 (m, 9h), 7.25 (s, 1h), 5.61 (s, 2h) ppm ; 13
c nmr (101 mhz, chloroform-d) δ 159.1, 153.5, 146.2, 142.1, 135.1, 134.2, 132.3, 130.8, 129.2, 128.0, 127.7, 126.7, 126.4, 115.4, 113.4, 95.7, 46.2 ppm;ir (kbr): v = 2919, 2850, 1625, 1494, 1453, 1356, 1190, 1059, 934, 834, 756, 699, 535 cm-1
.实施例46介离子杀虫剂v4的制备
向10 ml反应瓶中依次加入2-三氟苯基丙二酸二乙硫酯(134 mg, 0.40 mmol)、1 ml 1,3,5-三甲苯和n-(((2-氯噻唑-5-基)甲基)吡啶-2-胺(45 mg, 0.20 mmol),130 ℃ 搅拌2小时,tlc监测反应至完全。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得75 mg黄色晶状固体(收率85%)。
[0065]
1 (400 mhz, acetone-d6) δ 9.47 (d, j = 6.88 hz, 1h), 8.49
ꢀ–ꢀ
8.39 (m, 1h), 8.37 (s, 1h), 8.26 (d, j = 7.91 hz, 1h), 8.20 (d, j = 8.95 hz, 1h), 7.96 (s, 0h), 7.61 (t, j = 7.00 hz, 1h), 7.51 (dd, j = 19.12, 7.99 hz, 2h), 5.78 (s, 1h) ppm;
13
c nmr (101 mhz, acetone-d6) 158.7, 153.6, 152.5, 146.4, 144.1, 141.2, 136.8, 135.7, 134.1, 132.1, 128.7 (q, j= 30.3 hz), 127.0 (q, j = 4.15 hz), 125.0 (q, j = 272.7 hz), 121.6 (q, j = 3.95 hz), 116.5, 113.6, 92.1, 39.4 ppm.实施例47介离子杀虫剂v5的制备向10 ml反应瓶中依次加入2-(3,5-二氯苯基)丙二酸二乙硫酯(76 mg, 0.23 mmol)、1 ml 1,3,5-三甲苯和n-(((2-氯噻唑-5-基)甲基)吡啶-2-胺(26 mg, 0.11 mmol),130 ℃ 搅拌6小时。反应液冷却至室温,加入石油醚析出固体,过滤、干燥得23 mg黄色固体(收率46%)。1h nmr (400 mhz, chloroform-d) δ 9.54 (d, j = 6.85 hz, 2h), 8.20 (t, j = 7.98 hz, 1h), 7.77 (d, j = 1.84 hz, 2h), 7.67 (s, 1h), 7.60 (d, j = 8.95 hz, 1h), 7.44 (t, j = 7.01 hz, 1h), 5.59 (s, 2h) ppm; 13
c nmr (151 mhz, chloroform-d) δ 158.2, 154.6, 153.1, 145.8, 143.5, 140.5, 136.8, 134.0, 134.0, 133.0, 128.9, 126.4, 116.3, 112.1, 93.3, 40.0 ppm.最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管
参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1