一种羟氯喹粗品的精制方法与流程
本发明属于化学合成
技术领域:
,具体涉及一种羟氯喹粗品的精制方法。
背景技术:
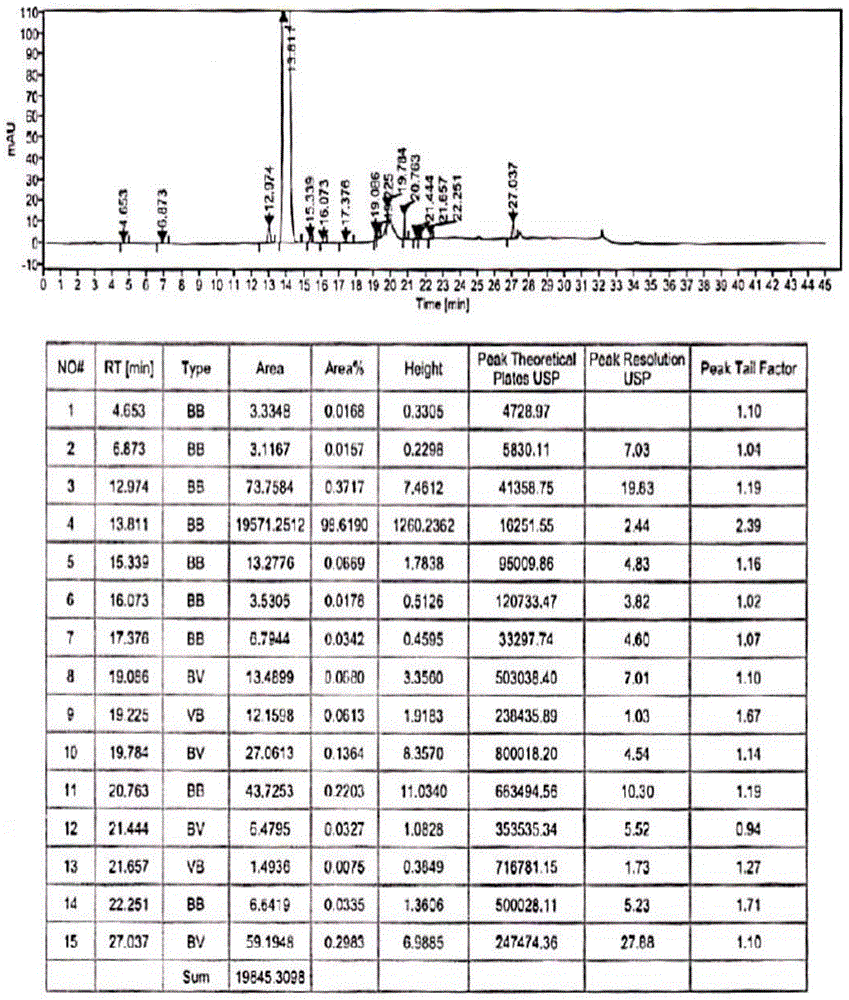
:硫酸羟氯喹(hydroxychloroquinesulfate,hcq),化学名:2-[[4-[(7-氯-4-喹啉基)氨基]戊基]乙氨基]-乙醇硫酸盐。由winthrop公司研制成功,于1956年在美国首次上市,1998年5月29日获得fda批准,用于治疗红斑狼疮、类风湿性关节炎,其化学结构如下:系统性红斑狼疮(systemiclupuserythematosus,sle)是一种多系统受累体内富含多种抗体的疾病,病因不清,治疗棘手。而抗疟药用于治sle始于1950年,dobois发现75%-80%的sle患者用抗疟药治疗后,皮疹、发热、关节症状改善,尤其是皮肤损害。其后研究发现,抗疟药可以使sle患者减少或停止使用皮质激素,并具有抗过敏作用。加拿大硫酸羟氯喹研究小组双盲对照研究表明,hcq能稳定sle患者的病情,明显减少复发。wallace等发现,hcq治疗可降低皮质激素依赖患者的血脂水平,可明显减少血栓形成的发生。鉴于hcq能为sle带来有益的作用,多数学者主张hcq用于轻到中度的sle患者,联合激素、免疫抑制剂用于重症sle的辅助治疗。随着科学家进一步研究发现,硫酸羟氯喹在风湿性疾病中可发挥多种免疫调节作用,首选硫酸羟氯喹具有显著的抗炎效应,它能稳定溶酶体、抑制酶的活性,进而抑制炎症介质的激活,同时可抑制炎症细胞如中性粒细胞的趋化和浸润,并显著减少促炎细胞因子如tnfα、il-l和il-6的产生:其次硫酸羟氯喹可通过抑制成纤维细胞的生长和结缔组织的沉积进而抑制关节炎患者滑膜的增生:再次硫酸羟氯喹可抑制抗原抗体的相互作用及免疫复合物的合成,促使类风湿因子滴度降低:硫酸羟氯喹还可通过影响紫外线吸收并阻挡紫外线对皮肤的伤害。以上种种皆为硫酸羟氯喹在临床治疗风湿性疾病提供大量依据。此外许多中心大规模临床研究也证实硫酸羟氯喹在风湿病的治疗中发挥显著的治疗效果。专利us2546658、cn102050781、wo2010027150、cn103724261、cn104230803等专利中公开制备硫酸羟氯喹的方法,以4,7-二氯喹啉与羟喹侧链为原料,先制备中间产物羟氯喹,然后,中间产物羟氯喹与硫酸成盐,即可得到硫酸羟氯喹。结果发现,在制备羟氯喹的反应过程中,有一脱乙基杂质(ep杂质c,ⅲ),结构式如下所示,用常规的重结晶方法均难去除,从而影响羟氯喹与硫酸成盐反应时,目标产物硫酸羟氯喹的收率和纯度。目前,在羟氯喹的精制过程中,杂质ⅲ使用溶剂乙酸乙酯、二氯甲烷及混合溶剂进行重结晶,均无明显效果,难以除去,从而影响硫酸羟氯喹的收率和纯度。专利ca2561987a1中公开了羟氯喹的精制方法,在精制的过程中使用了过量的衍生化试剂,使羟氯喹和杂质iii均发生衍生化反应,使羟氯喹和杂质均发生衍生化反应,再用氢氧化锂水解,最后重结晶,得到高纯度的羟氯喹,不仅操作繁琐,收率低,而且产生的废液较多,不适合工业化大生产。技术实现要素:本发明的目的是在现有技术的基础上,提供一种羟氯喹粗品的精制方法。本发明的技术方案如下:一种羟氯喹粗品的精制方法,它包括以下步骤:(1)一次精制:将羟氯喹粗品和乙酸乙酯搅拌混合,在搅拌的同时升温至60℃~80℃,待羟氯喹粗品完全溶解后,向其中加入衍生化试剂维持温度在60℃~80℃的条件下进行衍生化反应,待反应完成后,将得到的反应液在20℃~40℃的条件下进行搅拌析晶,再过滤,洗涤,干燥,得到一次精制品;其中:衍生化试剂为乙酸酐、丙酸酐、丁酸酐、异丁酸酐、boc酸酐、乙酰氯、丙酰氯、丁酰氯、氯苄或溴苄;衍生化试剂的质量为羟氯喹粗品总质量的0.5%~1.8%;羟氯喹粗品中包含杂质iii,该羟氯喹粗品的纯度为90.5%~99.5%,杂质iii的含量为0.2%~1.0%,其结构式如下:(2)二次精制:将步骤(1)中得到的一次精制品与和乙酸乙酯搅拌混合,在搅拌的同时升温至60℃~80℃,待一次精制品完全溶解后,将得到的混合溶液维持温度在20℃~40℃的条件下进行搅拌析晶,再过滤,洗涤,干燥,得到二次精制品。对于本发明而言,羟氯喹粗品包含杂质iii和其他杂质,例如,羟氯喹粗品的纯度为90.5%~99.5%,杂质iii的含量为0.2%~1.0%。本发明提供的羟氯喹粗品的精制方法,重点在于除去杂质iii。具体羟氯喹粗品中羟氯喹的纯度和杂质iii的含量可以根据实际情况进行选择,在一种优选方案中,羟氯喹粗品的纯度为97.5%~99.0%,杂质iii的含量为0.3%~0.5%。在羟氯喹粗品的制备过程中,得到的羟氯喹粗品中有效成分为羟氯喹(该化合物的结构式如式ii所示),具体合成路线如下,但不可避免也包含杂质iii和其他杂质,从而影响硫酸羟氯喹的收率和纯度。杂质iii与羟氯喹(化合物ii)的结构式很相似,采用简单的重结晶的方式难以除去。本发明是针对羟氯喹粗品中杂质iii难以除去的问题,提供一种除去杂质iii的方法,采用两次精制的过程,特别是在第一次精制的过程中,通过控制衍生化试剂的用量和衍生化反应的温度,使得羟氯喹粗品中的杂质ⅲ与衍生化试剂进行衍生化反应,将杂质ⅲ转移为杂质ⅳ、杂质v和杂质vi,基本上没有剩余的衍生化试剂与羟氯喹(化合物ii)生成杂质vii,再通过重结晶的方式除去杂质ⅳ、杂质v和杂质vi,有效成分羟氯喹几乎不参与衍生化反应,杂质vii的含量很低,在重结晶的过程中,不需要添加类似氢氧化锂这样的碱性金属氢氧化物,使得杂质vii水解成羟氯喹(化合物ii),达到改善羟氯喹(化合物ii)与过量衍生化试剂造成收率低的问题。在本发明中实现对羟氯喹粗品的精制,具体合成路线如下:其中,r为杂质iii与衍生化试剂进行衍生化反应时,引入的取代基,r具体指为哪一种取代基,取决于具体的衍生化试剂,可以但不局限于乙酰基、丙酰基、丁酰基、异丁酰基、叔丁氧羰基或苄基。对于本发明而言,在一次精制的过程中,主要是除去羟氯喹粗品中杂质iii,待搅拌析晶后,得到一次精制品,在一次精制品中几乎不存在杂质iii;再将得到的一次精制品与乙酸乙酯搅拌混合均匀,在20℃~40℃的条件下继续进行搅拌析晶,进一步除去其他的杂质,得到二次精制品,使得羟氯喹的纯度达到99.5%以上。在一种优选方案中,衍生化试剂为乙酸酐或丁酰氯,特别优选为乙酸酐。在步骤(1)中,在一次精制的过程中,需要严格控制衍生化试剂的用量,衍生化试剂的用量过低,无法除去羟氯喹粗品中的杂质iii,使得精制的效果不佳;而衍生化试剂的用量过高,不仅使得杂质iii与衍生化试剂进行衍生化反应,将杂质ⅲ转移为杂质ⅳ、杂质v和杂质vi,而且导致羟氯喹与过量较多的衍生化试剂进行衍生化反应生成杂质vii,使得羟氯喹粗品精制的过程中,羟氯喹大量损失,收率降低。在羟氯喹粗品精制的过程中又引入新的杂质vi,该杂质采用简单的重结晶方式难以除去,需要添加类似氢氧化锂这样的碱性金属氢氧化物,使得杂质vii水解成羟氯喹(化合物ii),才能解决羟氯喹收率低的难题,导致后处理过程繁琐,产生的废液较多,不适合工业化大生产。在步骤(1)中,在一次精制的过程中,衍生化试剂的质量为羟氯喹粗品总质量的0.5%~1.8%,可以但不局限于0.5%、0.55%、0.6%、0.65%、0.7%、0.8%、0.85%、0.9%、0.91%、0.95%、1.0%、1.1%、1.2%、1.22%、1.25%、1.27%、1.3%、1.35%、1.4%、1.45%、1.5%、1.52%、1.55%、1.6%、1.7%或1.8%,为了取得更好的效果,衍生化试剂的质量为羟氯喹粗品总质量的0.6%~1.6%。对本发明而言,发明人尝试了大量溶剂来溶解羟氯喹粗品,实现对羟氯喹粗品的一次精制,例如,溶剂可以但不局限于二氯甲烷、二氯乙烷、n,n-二甲基甲酰胺,n,n-二甲基乙酰胺,甲醇、乙醇、异丙醇和乙酸乙酯,结果发现:在相同的条件下,只有采用乙酸乙酯作为溶剂,在一次精制的过程中,杂质ⅲ几乎完全转移为杂质ⅳ、杂质v和杂质vi,在一次精制品中几乎未检测到杂质iii,而且羟氯喹的损失低,收率达到90%以上,纯度达到99.5%以上,其他单杂的含量低。而采用其他溶剂,在一次精制品中可以检测到杂质iii,而且杂质iii的含量较高,羟氯喹的收率低。在一次精制的过程中,羟氯喹粗品和乙酸乙酯的用量可以根据实际需要进行调整,在一种优选方案中,羟氯喹粗品与乙酸乙酯的质量体积比为4~15g/ml,可以但不局限于4g/ml、4.5g/ml、5g/ml、5.5g/ml、6g/ml、7g/ml、8g/ml、9g/ml、10g/ml、12g/ml、14g/ml或15g/ml,为了取得更好的效果,羟氯喹粗品与乙酸乙酯的质量体积比为5~10g/ml。在步骤(1)中,将羟氯喹粗品和乙酸乙酯搅拌混合,在搅拌的同时升温至60℃~80℃,可以但不局限于60℃、65℃、70℃、75℃或80℃,使得羟氯喹粗品完全溶解于乙酸乙酯中。在一种优选方案中,将羟氯喹粗品和乙酸乙酯搅拌混合,在搅拌的同时升温至65℃~75℃。对于本发明而言,在一次精制的过程中,需要严格控制衍生化反应的温度,使得杂质iii与衍生化试剂进行衍生化反应,将杂质ⅲ转移为杂质ⅳ、杂质v和杂质vi,而避免羟氯喹与衍生化试剂进行衍生化反应,导致羟氯喹大量损失。衍生化反应的温度过高或过低,降低了杂质iii与衍生化试剂进行衍生化反应时的竞争力,使得羟氯喹更容易与衍生化试剂进行衍生化反应生成杂质vii,导致羟氯喹大量损失,收率降低,又引入了新的杂质,该杂质采用简单的重结晶方式难以除去,导致后处理过程繁琐。在步骤(1)中,衍生化反应的温度为60℃~80℃,可以但不局限于60℃、65℃、70℃、75℃或80℃,为了取得更好的效果,衍生化反应的温度为65℃~75℃。进一步地,衍生化反应的时间为0.5~2小时,可以但不局限于0.5小时、1小时、1.5小时或2小时。在步骤(1)中,待衍生化反应结束后,将得到的反应液在20℃~40℃的条件下进行搅拌析晶,搅拌析晶的温度可以但不局限于20℃、25℃、30℃、35℃或40℃,为了取得更好的效果,搅拌析晶的温度优选为25℃~30℃。进一步地,搅拌析晶的时间为2~4小时,可以但不局限于2小时、2.5小时、3小时3.5小时或4小时.在步骤(1)中,待搅拌析晶后,将得到的混合溶液过滤、洗涤,干燥后,得到一次精制品。在一种优选方案中,洗涤时,采用乙酸乙酯进行洗涤。待洗涤结束后,干燥时,在40~60℃的条件下进行鼓风干燥,进一步优选地,干燥温度为50~55℃。在步骤(2)中,对一次精制品进一步精制时,将步骤(1)中得到的一次精制品和乙酸乙酯搅拌混合,在搅拌的同时升温至60℃~80℃,可以但不局限于60℃、65℃、70℃、75℃或80℃,使得一次精制品完全溶解于乙酸乙酯中。在一种优选方案中,将步骤(1)中得到的一次精制品和乙酸乙酯搅拌混合,在搅拌的同时升温至65℃~75℃。在步骤(2)中,在二次精制的过程中,一次精制品和乙酸乙酯的用量可以根据实际需要进行调整,在一种优选方案中,一次精制品与乙酸乙酯的质量体积比为4~10g/ml,可以但不局限于4g/ml、4.5g/ml、5g/ml、5.5g/ml、6g/ml、7g/ml、8g/ml、9g/ml或10g/ml为了取得更好的效果,一次精制品与乙酸乙酯的质量体积比为5g/ml。在步骤(2)中,待一次精制品完全溶解后,将得到的混合溶液维持温度在20℃~40℃的条件下进行搅拌析晶,搅拌析晶的温度可以但不局限于20℃、25℃、30℃、35℃或40℃,为了取得更好的效果,搅拌析晶的温度优选为20℃~25℃。进一步地,搅拌析晶的时间为2~4小时,可以但不局限于2小时、2.5小时、3小时3.5小时或4小时.在步骤(2)中,待搅拌析晶后,将得到的混合溶液过滤、洗涤,干燥后,得到二次精制品。在一种优选方案中,洗涤时,采用乙酸乙酯进行洗涤。待洗涤结束后,干燥时,在40~60℃的条件下进行鼓风干燥,进一步优选地,干燥温度为50~55℃。采用本发明的技术方案,优势如下:本发明针对羟氯喹粗品中杂质iii难以除去的问题,提供一种羟氯喹粗品的精制方法,包括两次精制过程,在第一次精制的过程中,通过控制衍生化试剂的用量和衍生化反应的温度,将杂质ⅲ转移为杂质ⅳ、杂质v和杂质vi,再通过重结晶的方式除去新杂质,而有效成分羟氯喹几乎不参与衍生化反应,实现对羟氯喹粗品的一次精制,在一次精制品中几乎不存在杂质iii,羟氯喹的损失低,收率达到90%以上,纯度达到99.5%以上。在第二次精制的过程中,采用简单的重结晶的方式,羟氯喹的收率高,收率达到95%左右,纯度达到99.9%。附图说明图1是实施例1中羟基氯喹粗品的hplc图;图2是实施例1中羟基氯喹一次精制品的hplc图;图3是实施例1中羟基氯喹二次精制品的hplc图。具体实施方式通过以下实施例并结合附图对本发明的羟氯喹的精制方法作进一步的说明,但这些实施例不对本发明构成任何限制。本发明羟基氯粗品及其精制品的检测方法如下:色谱条件:色谱柱由十八烷基硅烷键合硅胶为填充剂(4.6μm*25cm,5μm)的c18hplc柱;流动相组成为:流动相a:(乙腈-水-磷酸(体积比为10:90:0.2));流动相b:(乙腈-水-磷酸(体积比为80:20:0.1)),检测波长220nm,柱温为25℃,流速1.0ml/min,样品浓度0.2mg/ml,进样量20μl,按下表进行梯度洗脱。时间(min)a相(%)b相(%)010006100014851522010029010029.11000451000实施例1羟基氯喹粗品的精制(1)羟基氯喹粗品的制备:在1000ml三颈圆底烧瓶中,加入4,7-二氯喹啉100.02g,羟喹侧链132.07g和正丁醇200ml,控制温度为130~135℃,反应20小时至高效液相检测到达反应终点。将得到的反应液降温,加入400ml6%氢氧化钠水溶液,搅拌分相;水相再用100ml正丁醇萃取,合并有机相,用400ml水洗涤。有机相在50~65℃的条件下减压浓缩至无液体滴下。再加入500ml乙酸乙酯,5.00g活性炭,在60~70℃的条件下搅拌1小时。然后,趁热过滤,滤液自然降温至30℃,保温搅拌约12小时有大量固体析出后,再降低温度至10~20℃,保温搅拌3小时。过滤,乙酸乙酯洗涤,得到的固体在50~55℃的条件下鼓风干燥至恒重,得羟基氯喹粗品126.31g,收率74.48%,纯度98.62%,杂质iii含量为0.37%,具体色谱图见图1。(2)一次精制:在1000ml三颈圆底烧瓶中,加入步骤(1)中得到的125.00g羟氯喹粗品,625ml乙酸乙酯,在搅拌的过程中升温至68℃,待羟氯喹粗品完全溶解后,向其中加入0.76g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至25℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品121.26g,收率为97.01%,纯度为99.57%,杂质iii未检出,色谱图见图2。(3)二次精制:在1000ml三颈圆底烧瓶中,加入步骤(2)中得到的120.03g羟氯喹一次精制品,600ml乙酸乙酯,在搅拌的过程中升温至70℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至25℃,在此温度下保温搅拌析晶3小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品111.33g,收率为92.75%,纯度为99.81%,杂质iii未检出,其他最大单杂含量为0.05%,色谱图见图3。实施例2羟基氯喹粗品的精制(1)羟基氯喹粗品的制备:在5000ml三颈圆底烧瓶中,加入4,7-二氯喹啉500.00g,羟喹侧链660.05g和正丁醇1000ml,控制温度为130~135℃,反应20小时至高效液相检测到达反应终点。将得到的反应液降温,加入2000ml6%氢氧化钠水溶液,搅拌分相;水相再用500ml正丁醇萃取,合并有机相,用2000ml水洗涤。有机相在50~65℃的条件下减压浓缩至无液体滴下。再加入2500ml乙酸乙酯,25.00g活性炭,在60~70℃的条件下搅拌1小时。然后,趁热过滤,滤液自然降温至30℃,保温搅拌约12小时有大量固体析出后,再降低温度至10~20℃,保温搅拌3小时。过滤,乙酸乙酯洗涤,得到的固体在50~55℃的条件下鼓风干燥至恒重,得羟基氯喹粗品652.03g,收率76.89%,纯度98.54%,杂质iii含量为0.38%。(2)一次精制:在1000ml三颈圆底烧瓶中,加入步骤(1)中得到的100.02g羟氯喹粗品,1000ml乙酸乙酯,在搅拌的过程中升温至70℃,待羟氯喹粗品完全溶解后,向其中加入0.91g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至20℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品94.38g,收率为94.38%,纯度为99.67%,杂质iii未检出,其他最大单杂含量为0.18%。(3)二次精制:在1000ml三颈圆底烧瓶中,加入步骤(2)中得到的90.01g羟氯喹一次精制品,450ml乙酸乙酯,在搅拌的过程中升温至69℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌析晶3小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品85.14g,收率为94.60%,纯度为99.95%,杂质iii未检出,其他最大单杂含量为0.03%。实施例3羟基氯喹粗品的精制(1)一次精制:在1000ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的100g羟氯喹粗品,1000ml乙酸乙酯,在搅拌的过程中升温至71℃,待羟氯喹粗品完全溶解后,向其中加入1.22g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至30℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品94.12g,收率为94.12%,纯度为99.72%,杂质iii未检出,其他最大单杂含量为0.19%。(2)二次精制:在1000ml三颈圆底烧瓶中,加入步骤(1)中得到的90.02g羟氯喹一次精制品,600ml乙酸乙酯,在搅拌的过程中升温至70℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌析晶3小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品84.88g,收率为94.29%,纯度为99.95%,杂质iii未检出,其他最大单杂含量为0.02%。实施例4羟基氯喹粗品的精制(1)一次精制:在1000ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的100g羟氯喹粗品,1000ml乙酸乙酯,在搅拌的过程中升温至69℃,待羟氯喹粗品完全溶解后,向其中加入1.52g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至30℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品93.32g,收率为93.32%,纯度为99.65%,杂质iii未检出,其他最大单杂含量为0.21%。(2)二次精制:在1000ml三颈圆底烧瓶中,加入步骤(1)中得到的90.05g羟氯喹一次精制品,600ml乙酸乙酯,在搅拌的过程中升温至73℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌3析晶小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品85.68g,收率为95.15%,纯度为99.94%,杂质iii未检出,其他最大单杂含量为0.02%。对比例1羟基氯喹粗品的精制(1)一次精制:在250ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的10g羟氯喹粗品,100ml乙酸乙酯,在搅拌的过程中升温至71℃,待羟氯喹粗品完全溶解后,再降温至30℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品9.31g,收率为93.10%,纯度99.04%,杂质iii含量为0.32%。(2)二次精制:在100ml三颈圆底烧瓶中,加入步骤(1)中得到的9.05g羟氯喹一次精制品,45ml乙酸乙酯,在搅拌的过程中升温至72℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌析晶3小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品8.12g,收率为90.83%,纯度为99.77%,杂质iii含量为0.22%。对比例2羟基氯喹粗品的精制(1)一次精制:在250ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的10g羟氯喹粗品,100ml二氯甲烷,在搅拌的过程中升温至35℃,再降温至10℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品7.85g,收率为78.50%,纯度为99.61%,杂质iii含量为0.31%。(2)二次精制:在100ml三颈圆底烧瓶中,加入步骤(1)中得到的7.01g羟氯喹一次精制品,45ml二氯甲烷,在搅拌的过程中升温至回流,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至10℃,在此温度下保温搅拌析晶3小时,待析晶结束后,过滤,二氯甲烷洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品4.95g,收率为70.61%,纯度为99.71%,杂质iii含量为0.27%。对比例3羟基氯喹粗品的精制(1)一次精制:在250ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的10.02g羟氯喹粗品,100ml乙酸乙酯,在搅拌的过程中升温至71℃,待羟氯喹粗品完全溶解后,向其中加入0.30g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至20℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品9.12g,收率为91.20%。纯度为99.14%,杂质iii未检出,其他最大单杂含量为0.64%。(2)二次精制:在100ml三颈圆底烧瓶中,加入步骤(1)中得到的9.01g羟氯喹一次精制品,45ml乙酸乙酯,在搅拌的过程中升温至75℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌3析晶小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品7.81g,收率为86.68%,纯度为99.82%,杂质iii未检出,其他最大单杂含量为0.12%。对比例4羟基氯喹粗品的精制(1)一次精制:在250ml三颈圆底烧瓶中,加入实施例2步骤(1)中得到的10.05g羟氯喹粗品,100ml乙酸乙酯,在搅拌的过程中升温至55℃,羟氯喹粗品未完全溶解,向其中加入0.12g乙酸酐,在此温度下保温搅拌反应1小时,待反应结束后,将得到的反应液降温至20℃,在此温度下保温搅拌析晶3h,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹一次精制品9.34g,收率为92.94%。纯度为99.14%,杂质iii为0.21%,其他最大单杂含量为0.24%。(2)二次精制:在100ml三颈圆底烧瓶中,加入步骤(1)中得到的9.03g羟氯喹一次精制品,45ml乙酸乙酯,在搅拌的过程中升温至75℃,待羟氯喹一次精制品完全溶解后,将得到的混合溶液自然降温至20℃,在此温度下保温搅拌3析晶小时,待析晶结束后,过滤,乙酸乙酯洗涤,在50~55℃的条件下鼓风干燥至恒重,得到羟氯喹二次精制品8.25g,收率为91.56%,纯度为99.80%,杂质iii为0.20%,其他最大单杂含量为0.03%。以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可能对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1