一种D-π-A结构非线性化合物的制备方法及应用与流程
一种d
‑
π
‑
a结构非线性化合物的制备方法及应用
技术领域
1.本发明属于有机化学光学领域,具体涉及一种d
‑
π
‑
a结构非线性化合物的制备方法,涉及材料的制备方法及其性能测试。
背景技术:
2.香豆素及其衍生物是第一批在植物中发现的荧光染料。它们广泛分布于许多植物中,迄今已发现100多种天然香豆素化合物。其中,许多香豆素类型分子可以作为荧光染料、激光染料和光电材料,具有良好的稳定性和高荧光量子产率、大斯托克斯位移以及光物理和光化学性质可调、光稳定性好的性质。它们已成为荧光染料、光电分子设计中的首选荧光基团。同时,香豆素类化合物还具有一定的生物活性,广泛应用于生物医药领域。
3.从分子结构上看,香豆素类化合物由于内酯结构抑制了双键的旋转,从而提高了光稳定性。通过改变香豆素基上的给体
‑
受体部分取代基,控制分子内电荷转移,可以实现多种功能。香豆素分子结构中7位给电子基团、3位和4位吸电子基团的变化,使香豆素类化合物具有不同的荧光性质。香豆素类化合物的功能很大程度上取决于香豆素环上不同位置取代基的性质,尤其是3位和4位取代基的性质。通过合理设计,可以得到性能优异的光电分子。
4.而非线性光学材料,作为光电材料的一部分,在激光频率转换、四波混频、光束转向、图象放大、光信息处理、光存储、光纤通讯等领域具有越来越多的用途。由于香豆素优良的稳定性和高荧光量子产率、大斯托克斯位移以及通过不同取代基可调节的性质,本发明在此基础上进行进一步的合成,引入烷烃链同时添加不同基团改性,以期望得到更好的非线性光学性能,并对其中作为非线性双光子吸收的性能和作为双光子聚合引发剂的可能性进行了研究。
技术实现要素:
5.本发明的一个目的是解决至少上述问题和/或缺陷,并提供至少后面将说明的优点。
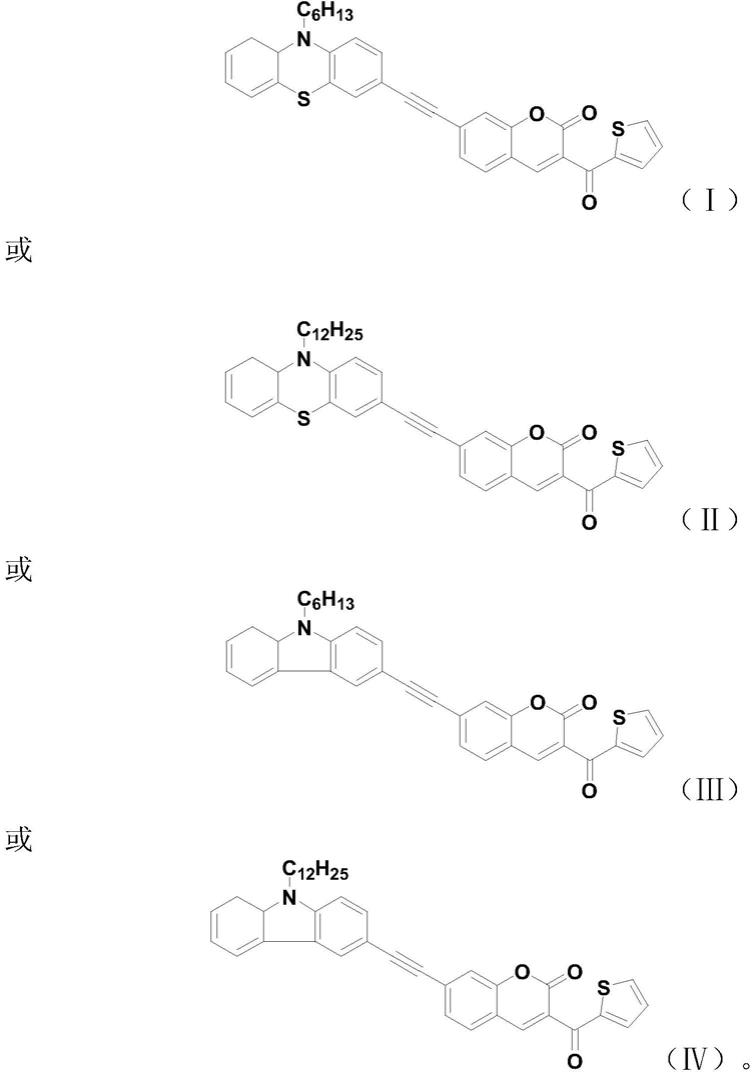
6.为了实现根据本发明的这些目的和其它优点,提供了一种d
‑
π
‑
a结构香豆素基非线性材料,其由香豆素基分子,通过碳碳三键作为共轭键桥,连接吩噻嗪或咔唑基团,构成d
‑
π
‑
a结构;其分子结构式为:
[0007][0008]
本发明还提供一种如上述的d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括如下步骤:
[0009]
步骤一、对原料4
‑
溴
‑2‑
羟基苯甲醛和3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯进行合成反应,生成产物溴
‑
香豆素;
[0010]
步骤二、对原料吩噻嗪或3
‑
溴
‑
9h
‑
咔唑通过取代反应加入己烷或十二烷基团;对吩噻嗪和nbs进行溴代反应,在活性位点引入卤代基团溴;
[0011]
步骤三、对步骤二所得产物进行sonogashira偶联反应,引入强极性炔羟基团;
[0012]
步骤四、对步骤三所得产物,在异丙醇中进行去保护,去除羟基;
[0013]
步骤五、对步骤一和步骤四所得产物进行sonogashira偶联反应,形成d
‑
π
‑
a结构化合物。
[0014]
优选的是,所述步骤一的过程为:将原料4
‑
溴
‑2‑
羟基苯甲醛加入去水去氧的三口瓶中,加入甲醇进行溶解,加入哌啶,回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯,磁力搅拌6个小时;反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素;
[0015]
所述4
‑
溴
‑2‑
羟基苯甲醛与3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯的摩尔比为1:1~1.2;所述4
‑
溴
‑2‑
羟基苯甲醛与甲醇的质量比为1g:10
‑
15ml。
[0016]
优选的是,所述步骤二的过程为:将nah缓慢加入冷却至0~3℃的含有吩噻嗪的dmf溶液中,20分钟后,缓慢加入1
‑
溴代正己烷或1
‑
溴代十二烷;将所得溶液在0~3℃下搅拌过夜,再缓慢加入去离子水;搅拌20分钟后,用乙酸乙酯萃取,然后用柱层析纯化,使用体积比为30~40:1的石油醚:乙酸乙酯为洗脱剂,得到10
‑
正己烷基
‑
10h
‑
吩噻嗪或10
‑
十二烷基
‑
10h
‑
吩噻嗪,为浅黄色油状物;
[0017]
向冷却至0~5℃的含有10
‑
正己烷基
‑
10h
‑
吩噻嗪或10
‑
十二烷基
‑
10h
‑
吩噻嗪的dcm溶液中缓慢加入nbs,将所得反应液在0~5℃下搅拌过夜,缓慢加入去离子水并搅拌30分钟,加入饱和氯化钠溶液萃取,得到有机相;然后使用硫酸镁干燥,过滤,除去溶剂,然后进行柱层析纯化,使用体积比为30~40:1的石油醚:乙酸乙酯为洗脱剂,得到3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3溴
‑
10
‑
十二烷基
‑
吩噻嗪;为浅黄色油状物;
[0018]
所述nah在贮存于矿物油中的质量分数为60%;所述nah与吩噻嗪的摩尔比为1.2~2:1;所述吩噻嗪与1
‑
溴代正己烷或1
‑
溴代十二烷的摩尔比为1:1~1.5;所述吩噻嗪与dmf的质量体积比为1g:6~10ml;所述10
‑
正己烷基
‑
10h
‑
吩噻嗪或10
‑
十二烷基
‑
10h
‑
吩噻嗪与nbs的摩尔比为1:1~1.2;所述10
‑
十二烷基
‑
10h
‑
吩噻嗪与dcm的摩尔体积比为1mmol:4ml;
[0019]
或所述步骤二的过程为:向冷却至0~5℃的含有3
‑
溴
‑
9h
‑
咔唑的dmf烧瓶中缓慢加入贮存于矿物油中的nah,10~30分钟后,缓慢加入1
‑
溴代正己烷或1
‑
溴代十二烷,将所得反应液在0~10℃下搅拌过夜并缓慢地加入去离子水,过滤并用石油醚洗涤后,得到3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑,为灰色固体;
[0020]
所述nah与3
‑
溴
‑
9h
‑
咔唑的摩尔比为1.2~2:1;所述3
‑
溴
‑
9h
‑
咔唑与1
‑
溴代正己烷或1
‑
溴代十二烷的摩尔比为1:0.8~1.5;所述3
‑
溴
‑
9h
‑
咔唑与dmf的质量体积比为1g:7.5~8.5ml。
[0021]
优选的是,所述步骤三的过程为:向反应器中加入3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪,然后加入cui、pdcl2(pph3)2、pph3、1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇和三乙胺,在ar保护下,使所得混合物在85~90℃下回流7~9小时,冷却后,使用减压蒸馏除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4~6:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇,为浅黄色油状物;
[0022]
或步骤三的过程为:向反应器中加入3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑,然后加入cui、pdcl2(pph3)2、pph3、1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇和三乙胺,在ar保护下,使所得混合物在85~90℃下回流7~9小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4~6:
1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇,为浅黄色油状物;
[0023]
所述3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪与cui的摩尔比为16~24:1;所述3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪与pdcl2(pph3)2的摩尔比为36~44:1;所述3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪与pph3的摩尔比为15~25:1;所述3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪与1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇的摩尔比为1:1.2~3;所述3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪或3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪与三乙胺的摩尔体积比为1mmol:6~10ml;
[0024]
所述3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑与cui的摩尔比为16~24:1;所述3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑与pdcl2(pph3)2的摩尔比为36~44:1;所述3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑与pph3的摩尔比为15~25:1;所述3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑与1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇的摩尔比为1:1.2~3;所述3
‑
溴
‑9‑
正己烷基
‑
咔唑或3
‑
溴
‑9‑
十二烷基
‑
咔唑与三乙胺的摩尔体积比为1mmol:6~10ml;
[0025]
优选的是,所述步骤四的过程为:对含有4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇的异丙醇溶液中加入氢氧化钾,在ar保护下,将所得混合物在85~90℃下回流2.5~4小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次50ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪为浅黄色油状物;
[0026]
所述4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇与氢氧化钾的摩尔比为1:3~5;所述4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇与异丙醇的摩尔体积比为5mmol:40~60ml;
[0027]
或所述步骤四中的过程为:对含有4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇的异丙醇溶液中加入氢氧化钾,在ar保护下,将所得混合物在85~90℃下回流2.5~4小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次50ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑,为浅黄色油状物。
[0028]
所述4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇与氢氧化钾的摩尔比为1:4~8;所述4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇或4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇与异丙醇的摩尔体积比为5mmol:40~60ml。
[0029]
优选的是,所述步骤五的过程为:向反应器中加入3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪,然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素、pdcl2(pph3)2、pph3、cui、三乙胺和dmf,并用ar脱气10~15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,
用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅰ)和分子结构式(ⅱ)的产物;
[0030]
所述3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与cui的摩尔比为16~24:1;所述3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与pdcl2(pph3)2的摩尔比为36~44:1;3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与pph3的摩尔比为15~25:1;所述3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与三乙胺的摩尔体积比为1mmol:10ml;所述3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与dmf的摩尔体积比为1mmol:5ml;所述3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪或3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪与7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素的摩尔比为1:1.1~1.5。
[0031]
或所述步骤五的过程为:向反应器中加入3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑,然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素、pdcl2(pph3)2、pph3、cui、三乙胺和dmf,并用ar脱气10~15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅲ)和分子结构式(ⅳ)的产物;
[0032]
所述3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与cui的摩尔比为16~24:1;所述3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与pdcl2(pph3)2的摩尔比为36~44:1;3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与pph3的摩尔比为15~25:1;所述3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与三乙胺的摩尔体积比为1mmol:10ml;所述3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与dmf的摩尔体积比为1mmol:5ml;所述3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑或3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑与7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素的摩尔比为1:1.1~1.5。
[0033]
本发明还提供一种如上述的d
‑
π
‑
a结构非线性化合物在树脂聚合中作为催化剂的应用,在5~30μmol/g的添加比例下,在飞秒激光的作用下,引发丙烯酸酯类树脂聚合,得到具有高固化率的交联体系。
[0034]
优选的是,所述丙烯酸酯类树脂为三羟甲基丙烷三丙烯酸酯或季戊四醇三丙烯酸酯。
[0035]
本发明还提供一种如上述的d
‑
π
‑
a结构非线性化合物在双光子聚合增材制造中的应用,其特征在于,利用nanoscribe gt设备进行双光子聚合增材制造,将d
‑
π
‑
a结构非线性化合物以摩尔比为10μmol/g的比例与三羟甲基丙烷三丙烯酸酯均匀混合搅拌,过滤,采用飞秒激光,重复频率为80mhz,波长为780
±
10nm;以5~50mw的激光功率,100
‑
100000μm/s的扫描速度进行增材制造,得到完整的三维结构。
[0036]
本发明至少包括以下有益效果:
[0037]
(1)本发明制备的几种d
‑
π
‑
a结构非线性材料,表现出良好的稳定性和多光子吸收性能,可用其进行双光子聚合反应,具有良好的性能。
[0038]
(2)本发明的d
‑
π
‑
a结构的香豆素基非线性材料的制备方法简便、提纯简单、所需时间短,得到的产物纯度高。
[0039]
(3)研究了本发明的非线性材料的结构和引发聚合性能,结果表明,该系列材料在50mw的激光功率下,聚合过程中扫描速度可达100,000μm/s。
[0040]
本发明的其它优点、目标和特征将部分通过下面的说明体现,部分还将通过对本
发明的研究和实践而为本领域的技术人员所理解。
附图说明:
[0041]
图1为本发明实施例1
‑
8中步骤一的化学反应式;
[0042]
图2为本发明实施例1和5中步骤二~步骤四的化学反应式;
[0043]
图3为本发明实施例1和5中步骤五的化学反应式
[0044]
图4为本发明实施例2和6中步骤五的化学反应式;
[0045]
图5为本发明实施例3和7中步骤二~步骤四的化学反应式;
[0046]
图6为本发明实施例3和7中步骤五的化学反应式;
[0047]
图7为本发明实施例4和8中步骤五的化学反应式;
[0048]
图8为本发明实施例1、5中产物的核磁氢谱图;
[0049]
图9为本发明实施例2、6中产物的核磁氢谱图;
[0050]
图10为本发明应用实施例1、5中产物引发聚合所成型结构的总体扫描电镜图。
[0051]
图11为本发明应用实施例3、7中产物引发聚合所成型结构的总体扫描电镜图。
具体实施方式:
[0052]
下面结合附图对本发明做进一步的详细说明,以令本领域技术人员参照说明书文字能够据以实施。
[0053]
应当理解,本文所使用的诸如“具有”、“包含”以及“包括”术语并不排除一个或多个其它元件或其组合的存在或添加。
[0054]
实施例1:
[0055]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0056]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(100ml)进行溶解,加入哌啶(2ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(1.98g,10mmol),磁力搅拌6个小时。反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.87,产率86%,纯度95%;
[0057]
步骤二、将nah(贮存于60%的矿物油中,3.00g,75mmol)缓慢加入冷却至0℃的含有吩噻嗪(10g,50mmol)的80ml dmf溶液中,20分钟后,缓慢加入1
‑
溴代正己烷(8.4ml,60mmol);将所得溶液在0℃下搅拌过夜,再缓慢加入去离子水;搅拌20分钟后,用乙酸乙酯萃取,然后用柱层析纯化,使用体积比为30:1的石油醚:乙酸乙酯为洗脱剂,得到10
‑
正己烷基
‑
10h
‑
吩噻嗪12.0g,产率85%,纯度96%,为浅黄色油状物;
[0058]
向冷却至0℃的含有10
‑
正己烷基
‑
10h
‑
吩噻嗪(5.66g,20mmol)的dcm溶液(80ml)中缓慢加入nbs(3.92g,22mmol),将所得反应液在0℃下搅拌过夜,缓慢加入去离子水并搅拌30分钟,加入饱和氯化钠溶液萃取,得到有机相;然后使用硫酸镁干燥,过滤,除去溶剂,然后进行柱层析纯化,使用体积比为30:1的石油醚:乙酸乙酯为洗脱剂,得到3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪6.44g,产率89%,纯度96%,为浅黄色油状物;
[0059]
步骤三、向反应器中加入3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪(3.63g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流7小时,冷却
后,使用减压蒸馏除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.52g,产率69%,纯度95%为浅黄色油状物;
[0060]
步骤四、对含有4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(1.83g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.112g,20mmol),在ar保护下,将所得混合物在90℃下回流2.5小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水(3
×
50ml)萃取,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪1.24g,产率81%,纯度95%为浅黄色油状物;
[0061]
步骤五、向反应器中加入3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪(307mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气10分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅰ)的产物359.7mg,产率64%,纯度95%,为橘黄色粉末。
[0062]
为了分析该合成材料的非线性吸收性能,进行了双光子聚合实验,使用不同功率的激光,在不同扫描速率下,研究了该实施例作为引发剂的光学性质和双光子聚合性能,如图11所示,结果表明,该引发剂可以引发聚合,聚合过程中扫描速度可达100,000μm/s。
[0063]
实施例2:
[0064]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0065]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(100ml)进行溶解,加入哌啶(2ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(1.98g,10mmol),磁力搅拌8个小时;反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.87g,产率86%,纯度95%;
[0066]
步骤二、将nah(贮存于60%的矿物油中,3.00g,75mmol)缓慢加入冷却至0℃的含有吩噻嗪(10g,50mmol)的80ml dmf溶液烧瓶中,并搅拌,25分钟后,缓慢加入1
‑
溴代十二烷(14.4ml,60mmol),将所得溶液搅拌12小时,再缓慢加入去离子水,搅拌半小时后,用乙酸乙酯萃取,然后用柱色谱法纯化,使用石油醚:乙酸乙酯=40:1(v:v)的洗脱剂,得到10
‑
十二烷基
‑
10h
‑
吩噻嗪,为浅黄色油状物15.06g,产率82%,纯度95%;
[0067]
向冷却至0℃的含有10
‑
十二烷基
‑
10h
‑
吩噻嗪(7.35g,20mmol)dcm溶液(80ml)中缓慢加入nbs(3.92g,22mmol),将所得反应液在0℃下搅拌过夜,缓慢加入去离子水并搅拌30分钟,加入饱和氯化钠溶液萃取,得到有机相;然后使用硫酸镁干燥,过滤,除去溶剂,然后进行柱层析纯化,使用体积比为30:1的石油醚:乙酸乙酯为洗脱剂,得到3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪8.21g,产率92%,纯度96%,为浅黄色油状物;
[0068]
步骤三、向反应器中加入3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪(4.46g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流9小时,冷却后,使用减压蒸馏除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次
100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4~6:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.92g,产率65%,纯度95%为浅黄色油状物;
[0069]
步骤四、对含有4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(2.25g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.390g,25mmol),在ar保护下,将所得混合物在85℃下回流4小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水(3
×
50ml)萃取,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪1.52g,产率78%,纯度95%为浅黄色油状物;
[0070]
步骤五、向反应器中加入3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪(392mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅱ)的产物400.5mg,产率62%,纯度95%,为橘黄色粉末。
[0071]
实施例3:
[0072]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0073]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(120ml)进行溶解,加入哌啶(3ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(2.376g,12mmol),磁力搅拌8个小时。反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.97g,产率89%,纯度95%;
[0074]
步骤二、向冷却至0℃的含有3
‑
溴
‑
9h
‑
咔唑(12.3g,50mmol)的dmf(100ml)烧瓶中缓慢加入贮存于矿物油中的nah((贮存于60%的矿物油中,3.00g,75mmol)),20分钟后,缓慢加入1
‑
溴代正己烷(10g,50mmol),将所得反应液在0℃下搅拌过夜并缓慢地加入去离子水,过滤并用石油醚洗涤后,得到3
‑
溴
‑9‑
正己烷基
‑
咔唑13.6g,产率83%,纯度95%。为灰色固体;
[0075]
步骤三、向反应器中加入3
‑
溴
‑9‑
正己烷基
‑
咔唑(3.29g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.79g,产率84%,纯度95%,为浅黄色油状物;
[0076]
步骤四、对含有4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(1.665g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.668g,30mmol),在ar保护下,将所得混合物在85℃下回流3小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次50ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑1.03g,产率73%,纯度95%,为浅黄色油状物;
[0077]
步骤五、向反应器中加入3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑(275mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气10~15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅲ)的产物333mg,产率63%,纯度95%,为橘黄色粉末。
[0078]
实施例4:
[0079]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0080]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(120ml)进行溶解,加入哌啶(3ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(2.376g,12mmol),磁力搅拌8个小时。反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.97g,产率89%,纯度95%;
[0081]
步骤二、向冷却至5℃的含有3
‑
溴
‑
9h
‑
咔唑(12.3g,50mmol)的dmf(80ml)烧瓶中缓慢加入贮存于矿物油中的nah(贮存于60%的矿物油中,3.00g,75mmol),30分钟后,缓慢加入1
‑
溴代十二烷(14.4ml,60mmol),将所得反应液在10℃下搅拌过夜并缓慢地加入去离子水,过滤并用石油醚洗涤后,得到3
‑
溴
‑9‑
十二烷基
‑
咔唑16.1g,产率78%,纯度95%;为灰色固体;
[0082]
步骤三、向反应器中加入3
‑
溴
‑9‑
十二烷基
‑
咔唑(4.14g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇3.06g,产率68%,纯度95%,为浅黄色油状物;
[0083]
步骤四、对含有4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(2.25g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.668g,30mmol),在ar保护下,将所得混合物在85℃下回流3小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次50ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑1.29g,产率66%,纯度95%,为浅黄色油状物;
[0084]
步骤五、向反应器中加入3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑(392mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气10~15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次30ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅳ)的产物333mg,产率63%,纯度95%,为橘黄色粉末。
[0085]
实施例5:
[0086]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0087]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加
入甲醇(120ml)进行溶解,加入哌啶(3ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(2.376g,12mmol),磁力搅拌6个小时。反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.97,产率89%,纯度95%;
[0088]
步骤二、将nah(贮存于60%的矿物油中,3.00g,75mmol)缓慢加入冷却至3℃的含有吩噻嗪(10g,50mmol)的80ml dmf溶液中,10分钟后,缓慢加入1
‑
溴代正己烷(10.5ml,75mmol);将所得溶液在0℃下搅拌过夜,再缓慢加入去离子水;搅拌20分钟后,用乙酸乙酯萃取,然后用柱层析纯化,使用体积比为40:1的石油醚:乙酸乙酯为洗脱剂,得到10
‑
正己烷基
‑
10h
‑
吩噻嗪11.4g,产率81%,纯度97%,为浅黄色油状物;
[0089]
向冷却至0~5℃的含有10
‑
正己烷基
‑
10h
‑
吩噻嗪(5.66g,20mmol)dcm溶液中缓慢加入nbs(3.92g,22mmol),将所得反应液在0℃下搅拌过夜,缓慢加入去离子水并搅拌10分钟,加入饱和氯化钠溶液萃取,得到有机相;然后使用硫酸镁干燥,过滤,除去溶剂,然后进行柱层析纯化,使用体积比为40:1的石油醚:乙酸乙酯为洗脱剂,得到3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪5.93g,产率82%,纯度96%,为浅黄色油状物;
[0090]
步骤三、向反应器中加入3
‑
溴
‑
10
‑
正己烷基
‑
吩噻嗪(3.63g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流9小时,冷却后,使用减压蒸馏除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为6:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.66g,产率73%,纯度96%为浅黄色油状物;
[0091]
步骤四、对含有4
‑
(10
‑
己基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(1.83g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.390g,25mmol),在ar保护下,将所得混合物在90℃下回流4小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水(3
×
50ml)萃取,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪1.14g,产率75%,纯度95%为浅黄色油状物;
[0092]
步骤五、向反应器中加入3
‑
乙炔基
‑
10
‑
己基
‑
10h
‑
吩噻嗪(307mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次40ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅰ)的产物381mg,产率68%,纯度95%,为橘黄色粉末。
[0093]
实施例6:
[0094]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0095]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(100ml)进行溶解,加入哌啶(2ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(1.98g,10mmol),磁力搅拌6个小时;反应物通过甲醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.97g,产率89%,纯度95%;
[0096]
步骤二、将nah(贮存于60%的矿物油中,3.00g,75mmol)缓慢加入冷却至0℃的含
有吩噻嗪(10g,50mmol)的80ml dmf溶液烧瓶中,并搅拌,25分钟后,缓慢加入1
‑
溴代十二烷(18ml,75mmol),将所得溶液搅拌12小时,再缓慢加入去离子水,搅拌半小时后,用乙酸乙酯萃取,然后用柱色谱法纯化,使用石油醚:乙酸乙酯=50:1(v:v)的洗脱剂,得到10
‑
十二烷基
‑
10h
‑
吩噻嗪,为浅黄色油状物16.16g,产率88%,纯度95%;
[0097]
向冷却至0~5℃的含有10
‑
十二烷基
‑
10h
‑
吩噻嗪(7.35g,20mmol)dcm溶液中缓慢加入nbs(3.92g,22mmol),将所得反应液在0℃下搅拌过夜,缓慢加入去离子水并搅拌30分钟,加入饱和氯化钠溶液萃取,得到有机相;然后使用硫酸镁干燥,过滤,除去溶剂,然后进行柱层析纯化,使用体积比为30:1的石油醚:乙酸乙酯为洗脱剂,得到3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪7.85g,产率88%,纯度95%,为浅黄色油状物;
[0098]
步骤三、向反应器中加入3
‑
溴
‑
10
‑
十二烷基
‑
吩噻嗪(4.46g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流9小时,冷却后,使用减压蒸馏除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次100ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.83g,产率63%,纯度97%为浅黄色油状物;
[0099]
步骤四、对含有4
‑
(10
‑
十二烷基
‑
10h
‑
吩噻嗪
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(2.25g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.668g,30mmol),在ar保护下,将所得混合物在85℃下回流2.5小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水(3
×
50ml)萃取,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪1.63g,产率84%,纯度96%为浅黄色油状物;
[0100]
步骤五、向反应器中加入3
‑
乙炔基
‑
10
‑
十二烷基
‑
10h
‑
吩噻嗪(392mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气15分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次40ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅱ)的产物432mg,产率67%,纯度96%,为橘黄色粉末。
[0101]
实施例7:
[0102]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0103]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(120ml)进行溶解,加入哌啶(2ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(2.376g,12mmol),磁力搅拌8个小时。反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.93g,产率85%,纯度96%;
[0104]
步骤二、向冷却至0~5℃的含有3
‑
溴
‑
9h
‑
咔唑(12.3g,50mmol)的dmf(100ml)烧瓶中缓慢加入贮存于矿物油中的nah((贮存于60%的矿物油中,3.60g,90mmol)),15分钟后,缓慢加入1
‑
溴代正己烷(10g,50mmol),将所得反应液在0℃下搅拌过夜并缓慢地加入去离子水,过滤并用石油醚洗涤后,得到3
‑
溴
‑9‑
正己烷基
‑
咔唑14.4g,产率88%,纯度95%;为灰色固体;
[0105]
步骤三、向反应器中加入3
‑
溴
‑9‑
正己烷基
‑
咔唑(3.29g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在90℃下回流9小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次80ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为4:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.59g,产率78%,纯度96%,为浅黄色油状物;
[0106]
步骤四、对含有4
‑
(9
‑
己基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(1.665g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.668g,30mmol),在ar保护下,将所得混合物在90℃下回流3小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次60ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑1.10g,产率78%,纯度97%,为浅黄色油状物;
[0107]
步骤五、向反应器中加入3
‑
乙炔基
‑9‑
己基
‑
9h
‑
咔唑(275mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气10分钟,使所得混合物回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次50ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅲ)的产物348mg,产率66%,纯度96%,为橘黄色粉末。
[0108]
实施例8:
[0109]
一种d
‑
π
‑
a结构香豆素基非线性材料的制备方法,包括以下步骤:
[0110]
步骤一、将原料4
‑
溴
‑2‑
羟基苯甲醛(2.01g,10mmol)加入去水去氧的三口瓶中,加入甲醇(80ml)进行溶解,加入哌啶(2ml),回流,使用恒压滴液漏斗缓慢加入3
‑
氧代
‑3‑
噻吩
‑2‑
基丙酸乙酯(2.376g,12mmol),磁力搅拌6个小时;反应物用乙醇进行重结晶,得到白色片状物7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素2.86g,产率86%,纯度96%;
[0111]
步骤二、向冷却至0℃的含有3
‑
溴
‑
9h
‑
咔唑(12.3g,50mmol)的dmf(80ml)烧瓶中缓慢加入贮存于矿物油中的nah(贮存于60%的矿物油中,3.00g,75mmol),30分钟后,缓慢加入1
‑
溴代十二烷(14.4ml,60mmol),将所得反应液在5℃下搅拌过夜并缓慢地加入去离子水,过滤并用石油醚洗涤后,得到3
‑
溴
‑9‑
十二烷基
‑
咔唑17.1g,产率83%,纯度95%;为灰色固体;
[0112]
步骤三、向反应器中加入3
‑
溴
‑9‑
十二烷基
‑
咔唑(4.14g,10mmol),cui(95mg,0.5mmol),pdcl2(pph3)2(175mg,0.25mmol),pph3(131mg,0.5mmol),1,1
‑
二甲基
‑2‑
丁炔
‑1‑
醇(1.45ml,15mmol)和三乙胺(80ml),在ar保护下,使所得混合物在85℃下回流8小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次80ml,合并有机层,用mgso4干燥,浓缩,并使用体积比为6:1的石油醚和乙酸乙酯作为洗脱剂,通过柱色谱法纯化,得到4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇2.88g,产率68%,纯度97%,为浅黄色油状物;
[0113]
步骤四、对含有4
‑
(9
‑
十二烷基
‑
9h
‑
咔唑
‑3‑
基)
‑2‑
甲基
‑3‑
炔
‑2‑
醇(2.25g,5mmol)的异丙醇溶液(50ml)中加入氢氧化钾(1.668g,30mmol),在ar保护下,将所得混合物
在85℃下回流4小时,反应完成后,在保持通氩气的同时冷却,浓缩后,溶于乙酸乙酯,用饱和食盐水萃取3次,每次60ml,用无水硫酸镁干燥并浓缩,深棕色残余物通过柱色谱法纯化,用体积比为20:1的石油醚和乙酸乙酯作为洗脱剂,得到3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑1.21g,产率62%,纯度95%,为浅黄色油状物;
[0114]
步骤五、向反应器中加入3
‑
乙炔基
‑9‑
十二烷基
‑
9h
‑
咔唑(392mg,1mmol),然后加入7
‑
溴
‑3‑
(噻吩
‑2‑
羰基)
‑
香豆素(402mg,1.2mmol)、cui(9.5mg,0.05mmol),pdcl2(pph3)2(17.6mg,0.025mmol),pph3(13.1mg,0.05mmol),和三乙胺(10ml),dmf(5ml),并用ar脱气10分钟,使所得混合物回流9小时,冷却后,除去溶剂,将残余物倒入饱和食盐水中并用乙酸乙酯萃取3次,每次50ml,合并有机层,用mgso4干燥,浓缩并通过柱色谱法纯化,得到分子结构式(ⅳ)的产物322mg,产率61%,纯度95%,为橘黄色粉末。
[0115]
实施例9:
[0116]
一种实施例1制备的d
‑
π
‑
a结构非线性化合物在树脂聚合中作为催化剂的应用,在10μmol/g的添加比例下,在飞秒激光的作用下,引发三羟甲基丙烷三丙烯酸酯聚合,得到具有高固化率的交联体系。
[0117]
实施例10:
[0118]
一种实施例2制备的d
‑
π
‑
a结构非线性化合物在树脂聚合中作为催化剂的应用,在10μmol/g的添加比例下,在飞秒激光的作用下,引发三羟甲基丙烷三丙烯酸酯聚合,得到具有高固化率的交联体系。
[0119]
实施例11:
[0120]
一种实施例3制备的d
‑
π
‑
a结构非线性化合物在树脂聚合中作为催化剂的应用,在10μmol/g的添加比例下,在飞秒激光的作用下,引发三羟甲基丙烷三丙烯酸酯聚合,得到具有高固化率的交联体系。
[0121]
实施例12:
[0122]
一种实施例4制备的d
‑
π
‑
a结构非线性化合物在树脂聚合中作为催化剂的应用,在10μmol/g的添加比例下,在飞秒激光的作用下,引发三羟甲基丙烷三丙烯酸酯聚合,得到具有高固化率的交联体系。
[0123]
实施例13:
[0124]
一种实施例1制备的d
‑
π
‑
a结构非线性化合物在双光子聚合增材制造中的应用,利用nanoscribe gt设备进行双光子聚合增材制造,将d
‑
π
‑
a结构非线性化合物以摩尔比为10μmol/g的比例与三羟甲基丙烷三丙烯酸酯均匀混合搅拌,过滤,采用飞秒激光,重复频率为80mhz,波长为780
±
10nm;以50mw的激光功率,100000μm/s的扫描速度进行增材制造,得到完整的三维结构。
[0125]
实施例14:
[0126]
一种实施例2制备的d
‑
π
‑
a结构非线性化合物在双光子聚合增材制造中的应用,利用nanoscribe gt设备进行双光子聚合增材制造,将d
‑
π
‑
a结构非线性化合物以摩尔比为10μmol/g的比例与三羟甲基丙烷三丙烯酸酯均匀混合搅拌,过滤,采用飞秒激光,重复频率为80mhz,波长为780
±
10nm;以50mw的激光功率,100000μm/s的扫描速度进行增材制造,得到完整的三维结构。
[0127]
实施例15:
[0128]
一种实施例3制备的d
‑
π
‑
a结构非线性化合物在双光子聚合增材制造中的应用,利用nanoscribe gt设备进行双光子聚合增材制造,将d
‑
π
‑
a结构非线性化合物以摩尔比为10μmol/g的比例与三羟甲基丙烷三丙烯酸酯均匀混合搅拌,过滤,采用飞秒激光,重复频率为80mhz,波长为780
±
10nm;以50mw的激光功率,100000μm/s的扫描速度进行增材制造,得到完整的三维结构。
[0129]
实施例16:
[0130]
一种实施例4制备的d
‑
π
‑
a结构非线性化合物在双光子聚合增材制造中的应用,利用nanoscribe gt设备进行双光子聚合增材制造,将d
‑
π
‑
a结构非线性化合物以摩尔比为10μmol/g的比例与三羟甲基丙烷三丙烯酸酯均匀混合搅拌,过滤,采用飞秒激光,重复频率为80mhz,波长为780
±
10nm;以50mw的激光功率,100000μm/s的扫描速度进行增材制造,得到完整的三维结构。尽管本发明的实施方案已公开如上,但其并不仅仅限于说明书和实施方式中所列运用,它完全可以被适用于各种适合本发明的领域,对于熟悉本领域的人员而言,可容易地实现另外的修改,因此在不背离权利要求及等同范围所限定的一般概念下,本发明并不限于特定的细节和这里示出与描述的图例。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1