一种1,2,3-三氮唑衍生物及其制备方法与应用
本公开属于有机合成化学技术领域,具体提供一种1,2,3-三氮唑衍生物及其制备方法与应用。
背景技术:
这里的陈述仅提供与本公开有关的背景信息,而不必然构成现有技术。
1,2,3-三氮唑是一类由三个氮原子和两个碳原子构建的五元芳杂环化合物。1,2,3-三氮唑化合物有着独特的化学结构性质,是构建杂环化合物的一种重要结构单元,并且可以利用炔烃和叠氮引入不同的取代基。1,2,3-三氮唑化合物生理活性丰富,易于制备,稳定性强,广泛应用于各大领域。在生物医药方面,1,2,3-三氮唑是一种广为人知的结构单元骨架,广泛存在于许多生物化合物中。其中取代基的立体结构、空间位阻、电子效应能通过改变三唑环上的电子密度,从而使其表现出多样的生物性能;在材料领域中,1,2,3-三氮唑在化学传感器领域应用也是相当广泛;在有机合成领域中,1,2,3-三氮唑因为特殊的母体结构为很多金属配体提供了多个供体位点,因此可以作为很重要的生物药物合成中间体。所以,三氮唑类化合物官能团的改进引起了人们的广泛探索。
1,2,3-三氮唑最早是由michael发现的,1893年报道了富含氮原子的三唑环结构,但他并未进行更深一步的研究。直到1963年,huisgen深入研究端炔和叠氮化合物发生1,3-偶极环加成反应,得到多取代三氮唑,此反应确定为huisgen反应。然而,该反应的选择性较差,同时获得了1,4-二取代-1,2,3-三氮唑和1,5-二取代-1,2,3-三氮唑,且对温度要求较高,需加热至100℃以上。2002年,shapless和他的团队意外发现cu(i)可以促进端炔和叠氮化合物通过1,3-偶极环加成反应制备1,4-二取代-1,2,3-三氮唑。该反应操作简单、产率高、反应条件温和、具有高度立体专一性和区域选择性,该反应被命名为cuaac反应。近几年来,对1,2,3-三氮唑的研究更是层出不穷。2014年,jabeenakhazir等人以parthenin为起始原料,采用点击化学方法设计合成了一系列1,2,3-三唑类同系物,并对其对6种人类肿瘤细胞系(pc-3、thp-1、hct-15、hela、a-549和mcf-7)的细胞毒性进行了评价;2016年,mohammadmahdavi课题组研究了以靛酸酐为起始原料,经三步反应合成2,3-二氢喹唑啉-4(1h)-酮的新型1,2,3-三唑衍生物的有效途径。由靛酸酐和苄胺反应得到的2-氨基-n-取代苯甲酰胺与苯甲醛进行偶联环化反应,然后与原位合成的有机叠氮化合物进行点击反应,得到了产率较高的标题化合物;2019年,martinatireli等人设计、合成了一系列以亚油酸为基础的1,2,3-三唑衍生物,并进行了基于细胞的nf-κb抑制筛选实验。在被测化合物中,化合物6k表现出显著的nf-κb抑制活性,ic50值在低微摩尔范围内。分子对接研究揭示了6k与nf-κb之间的关键相互作用,其中1,2,3-三唑部分和aa骨架的羟基对提高抑制活性非常重要;2020年,aysetan等人研究了采用铜催化叠氮炔环加成反应合成了含糖骨架的1,2,3-三唑类化合物,并考察了它们对黄嘌呤氧化酶的体外抑制作用。
然而,在本公开发明人在研究过程中发现,实现官能团化1,2,3-三氮唑化合物的方法中存在以下问题:(1)所用催化剂价格昂贵,所需成本较高。(2)反应条件繁琐,难以控制。(3)反应步骤多,收率低。(4)合成的1,2,3-三唑类化合物官能团单一,在药物合成中不能发挥其优势。
技术实现要素:
为了解决现有技术中1,2,3-三氮唑衍生物合成产物官能团较为单一,难以充分发挥其在药物化学中的优势;同时合成方法步骤繁琐,原料价格较贵,产率较低的问题。同时发挥多种官能团在1,2,3-三氮唑中的作用,保护基团和产物,本公开首次实现了在此类化合物中引入三种官能团,同时能够利用端炔、酚取代的烯基叠氮化合物、酯类化合物一锅法实现官能团化1,2,3-三氮唑衍生物的方法。
为了实现上述目的,本公开的技术方案为:
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,其结构如式(1)所示,
其中,a1选自氰基、羰甲基、羰丙基;a2选自h、醚基、c1-c8直链或支链的烷基;
b选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c1-c8直链或支链的烷基,c1-c2直链或支链烷氧基;
优选的,b选自苯基、
本公开一个或一些实施方式中,提供一种合成式(1)所示化合物的方法,其中,所述方法包括以端炔、酚取代的烯基叠氮化合物、酯类化合物作为原料,在一价铜盐和二价钯盐的催化作用下,通过一锅法合成式(1)所示化合物;
其中,所述端炔具有
r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c1-c8直链或支链的烷基,c1-c2直链或支链烷氧基;
其中,a1、a2、b同权利要求1
其中,酚取代的烯基叠氮化合物具有式iii所示结构;
其中,酯类化合物选自
所述方法按如下所示的反应路线进行:
上述技术方案中的一个或一些技术方案具有如下优点或有益效果:
1)本公开首次提供了一种双金属催化端炔实现官能团化1,2,3-三氮唑衍生物的方法,丰富了三氮唑类化合物的药物性质,充分发挥了其在药物合成化学领域中的优势。该方法简便、高效,所用到的原料及催化剂易得且无毒、步骤少、成本低、条件温和可控,本公开提供的方法适宜大规模生产。
2)本公开首次以端炔、酚取代的烯基叠氮化合物、酯类化合物作为原料,在一价铜盐和二价钯盐的催化作用下,通过一锅法合成结构对称式1,2,3-三氮唑衍生物的方法。此类化合物引入了氰基,丙烯基和酯基三种官能团,化学性质稳定,应用范围较广,在药物合成中意义重大。同时,本公开的方法成本较低,操作简单,条件易于控制。
附图说明
构成本公开一部分的说明书附图用来提供对本公开的进一步理解,本公开的示意性实施例及其说明用于解释本公开,并不构成对本公开的不当限定。
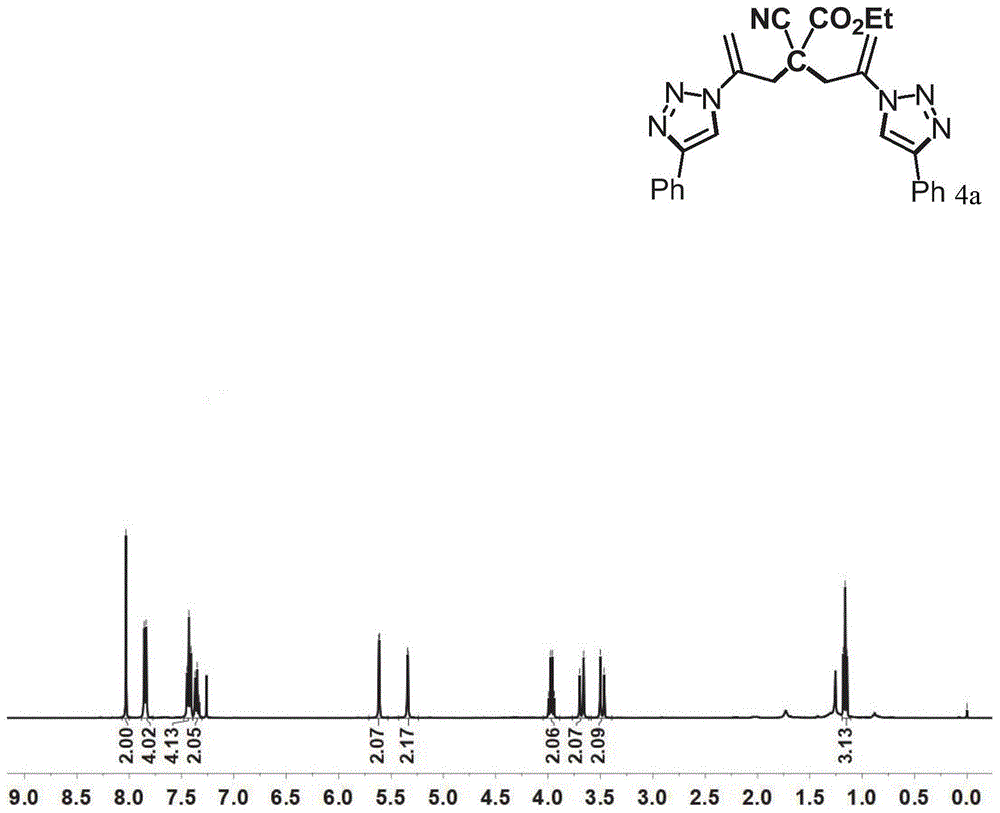
图1为本公开实施例9制备的化合物4a的1h-nmr的核磁共振谱图;
图2为本公开实施例9制备的化合物4a的13c-nmr的核磁共振谱图;
图3为本公开实施例10制备的化合物4b的1h-nmr的核磁共振谱图;
图4为本公开实施例10制备的化合物4b的13c-nmr的核磁共振谱图;
图5为本公开实施例11制备的化合物4c的1h-nmr的核磁共振谱图;
图6为本公开实施例11制备的化合物4c的13c-nmr的核磁共振谱图;
图7为本公开实施例12制备的化合物4d的1h-nmr的核磁共振谱图;
图8为本公开实施例12制备的化合物4d的13c-nmr的核磁共振谱图;
图9为本公开实施例13制备的化合物4e的1h-nmr的核磁共振谱图;
图10为本公开实施例13制备的化合物4e的13c-nmr的核磁共振谱图;
图11为本公开实施例14制备的化合物4f的1h-nmr的核磁共振谱图;
图12为本公开实施例14制备的化合物4f的13c-nmr的核磁共振谱图;
图13为本公开实施例15制备的化合物4g的1h-nmr的核磁共振谱图;
图14为本公开实施例15制备的化合物4g的13c-nmr的核磁共振谱图。
具体实施方式
下面将对本公开实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅是本公开的一部分实施例,而不是全部实施例。基于本公开的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其它实施例,都属于本公开保护的范围。
为了解决现有技术中1,2,3-三氮唑衍生物合成产物官能团较为单一,难以充分发挥其在药物化学中的优势;同时合成方法步骤繁琐,原料价格较贵,产率较低的问题。同时发挥多种官能团在1,2,3-三氮唑中的作用,保护基团和产物,本公开首次实现了在此类化合物中引入三种官能团,同时能够利用端炔、酚取代的烯基叠氮化合物、酯类化合物一锅法实现官能团化1,2,3-三氮唑衍生物的方法。
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,其结构如式(1)所示,
其中,a1选自氰基、羰甲基、羰丙基;a2选自h、醚基、c1-c8直链或支链的烷基;
b选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c1-c8直链或支链的烷基,c1-c2直链或支链烷氧基;
优选的,b选自苯基、
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,其结构如式(2),式(3),式(4)所示,
其中,b选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c1-c8直链或支链的烷基,c1-c2直链或支链烷氧基;
优选的,b选自苯基、
y选自h、c1-c8直链或支链的烷基、醚基。
本公开一个或一些实施方式中,提供一种1,2,3-三氮唑衍生物,选自以下化合物:
本公开一个或一些实施方式中,提供一种合成式(1)所示化合物的方法,其中,所述方法包括以端炔、酚取代的烯基叠氮化合物、酯类化合物作为原料,在一价铜盐和二价钯盐的催化作用下,通过一锅法合成式(1)所示化合物;
其中,所述端炔具有
r选自芳基、给电子或吸电子的取代芳基、给电子或吸电子的杂芳基,c1-c8直链或支链的烷基,c1-c2直链或支链烷氧基;
其中,a1、a2、b同权利要求1
其中,酚取代的烯基叠氮化合物具有式iii所示结构;
其中,酯类化合物选自
所述方法按如下所示的反应路线进行:
优选的,一价铜盐为cui、cutc、cu2s、cu2o、cucl中的一种;
或,二价钯盐为pd(pph3)4、pd(pph3)2cl2、pd(oac)2中的一种;
所述催化剂能够提高原料的转化率和产物的产率。
该实施方式的一种或多种实施例中,当一价铜盐为碘化亚铜时,二价钯盐为pd(pph3)4时,能够进一步提高该1,2,3-三氮唑衍生物的产率。
或,添加剂为三乙胺、dbu、naoh、pmdeta中的一种或多种;当添加物为三乙胺时,能够提高原料的转化率和产物的产率。
或,溶剂为二甲亚砜(dmso)、乙腈(ch3cn)、n,n-二甲基甲酰胺(dmf)、甲苯、1,2-二氯乙烷(dce)、甲醇中的一种。该溶剂提高原料的转化率,同时提高产物的产率。当溶剂为乙腈时,原料的转化率、产物的产率更高。
优选的,所述芳基选自苯基和取代苯基;
进一步地,所述芳基选自苯基和被卤素、烷基或烷氧基所取代的苯基;
进一步地,所述卤素选自f、cl、br;
进一步地,所述烷基选自c1-c8直链或支链烷基;
进一步地,所述c1-c8直链烷基选自甲基、乙基、正丙基,正丁基;
进一步地,所述c1-c8支链烷基选自叔丁基、正戊基;
进一步地,所述烷氧基选自c1-c2直链或支链烷氧基;
进一步地,所述c1-c2直链或支链烷氧基选自甲氧基、乙氧基等。
进一步地,所述杂芳基含有一个或多个杂原子,所述杂原子选自n、o和s;
具体的,r选自正丁基、正戊基、苯基、4-乙氧基苯基、4-氟苯基。
优选的,端炔、酚取代的烯基叠氮化合物、酯类化合物的摩尔比为1~3:1~8:1~7;
优选的,端炔、酚取代的烯基叠氮化合物、酯类化合物的摩尔比为1:7:6;
或,一价铜盐的添加量为原料总质量的10%~50%,二价钯盐的添加量为原料总质量的10%;
优选的,一价铜盐的添加量为原料总质量的30%,二价钯盐的添加量为原料总质量的2%;
或,反应时间为0~6h,反应时间不为0;
优选的,反应时间为4±0.5h;
或,反应温度为50~100℃;优选为70±6℃。选择该温度范围能够进一步提高原料的转化率和产物的产率。
优选的,还包括提纯过程,所述提纯过程包括如下步骤:将反应后溶液加入萃取溶剂进行萃取获得有机相,将有机相中的溶剂去除,进行硅胶柱层析,获得纯度更高的化合物;
优选的,萃取采用的萃取溶剂为1,2-二氯乙烷、甲苯、乙酸乙酯、乙醚、正己烷、环己烷、石油醚或二氯甲烷中的一种或多种;
进一步优选的,萃取采用的萃取溶剂为二氯甲烷;
优选的,所述萃取进行1~3次,每次使用5~20ml萃取溶剂;
优选的,获得有机相采用无水硫酸镁进行干燥,再去除有机溶剂;
优选的,硅胶柱层析的洗脱液为石油醚和乙酸乙酯;
优选的,石油醚和乙酸乙酯的体积比为1~30:1~6;
优选的,石油醚和乙酸乙酯的体积比为2:1。采用该洗脱液能够获得纯度更高的1,2,3-三氮唑衍生物。
优选的,为了使端炔、酚取代的烯基叠氮化合物、酯类化合物混合均匀,该实施方式的一种或多种实施例中,反应包括如下步骤:将原料添加至溶剂中溶解,在加入添加剂及催化剂的作用下,加热进行反应。
本公开一个或一些实施方式中,提供上述1,2,3-三氮唑衍生物或上述方法制得的化合物在制备抗炎药物中的应用。
实施例1
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入cutc(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为46%。
实施例2
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入氯化亚铜(0.0108g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为40%。
实施例3
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的dmf中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.0075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为55%。
实施例4
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的甲苯中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为60%。
实施例5
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为91%。
实施例6
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)三乙胺(0.0700ml,2.5mmol),加入到1ml的dce中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为65%。
实施例7
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、醋酸钯(0.0011g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为72%。
实施例8
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、氢氧化钠(0.0470ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0062g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为47%。
实施例9
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、pmdeta(0.5220ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4a的产率为58%。
实施例1~9的反应如下所示:
实施例10
将化合物1b即4-乙炔基苯甲酸甲酯(0.0390ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4b的产率为87%。
反应如下所示:
化合物4b:
1hnmr(400mhz,cdcl3),如图3所示,δ8.15(s,2h),7.97(d,j=8.2hz,4h),7.72(d,j=8.2hz,4h),5.65(d,j=2.4hz,2h),5.40(d,j=2.3hz,2h),4.12(q,j=7.1hz,1h),4.04(q,j=7.2hz,2h),3.68(d,j=14.8hz,2h),3.52(d,j=14.8hz,2h),2.05(s,2h),1.24(d,j=19.9,7.1hz,6h).hrms(esi)m/zcalculatedforc31h29n7o6[m+na]+:618.2098,found:618.2068.13cnmr(100mhz,cdcl3),如图4所示,δ166.59,146.45,137.57,134.13,132.80,126.31,119.24,118.61,116.24,112.05,110.84,63.89,53.46,48.71,39.04,13.81.
实施例11
将化合物1c即4-乙基苯乙炔(0.0330ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4c的产率为85%。
反应如下所示:
化合物4c:
1hnmr(400mhz,cdcl3),如图5所示,δ7.99(s,1h),7.89–7.70(m,2h),7.39–7.18(m,2h),5.60(d,j=2.3hz,1h),5.33(d,j=2.3hz,1h),3.95(q,j=7.2hz,1h),3.68(d,j=14.8hz,1h),3.47(d,j=14.9hz,1h),2.68(q,j=7.6hz,2h),1.26(t,j=7.6hz,4h),1.15(t,j=7.1hz,2h).hrms(esi)m/zcalculatedforc31h33n2o2[m+na]+:558.2598,found:558.2576.13cnmr(100mhz,cdcl3),如图6所示,δ166.62,148.34,144.88,137.70,128.41,127.25,125.92,117.58,116.66,109.81,63.75,48.91,39.04,28.71,15.51,13.72.
实施例12
将化合物1d即1-庚炔(0.0340ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3a即氰乙酸乙酯(0.0330ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4d的产率为90%。
反应如下所示:
化合物4d:
1hnmr(400mhz,cdcl3),如图7所示,δ7.54(s,2h),5.49(d,j=2.1hz,2h),5.25(d,j=2.1hz,2h),3.97(q,j=7.2hz,2h),3.61(d,j=14.8hz,2h),3.41(d,j=14.8hz,2h),2.77–2.66(m,4h),1.69(q,j=7.5hz,4h),1.30-1.35(m,8h),1.20(t,j=7.2hz,3h).hrms(esi)m/zcalculatedforc25h37n7o2[m+na]+:490.2898,found:490.2834.13cnmr(100mhz,cdcl3),如图8所示,δ166.62,149.09,137.78,119.11,116.55,109.07,63.51,48.82,39.00,31.36,28.97,25.52,22.39,13.99,13.74.
实施例13
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3b即乙酰乙酸甲氧乙酯(0.0370ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4e的产率为83%。
反应如下所示:
化合物4e:
1hnmr(400mhz,cdcl3),如图9所示,δ7.92(s,2h),7.86–7.79(m,4h),7.42(t,j=7.5hz,4h),7.34(t,j=8.5,6.2hz,2h),5.50(d,j=1.7hz,2h),5.19(d,j=1.8hz,2h),4.15–4.04(m,2h),3.61(d,j=15.8hz,2h),3.55–3.40(m,4h),3.37–3.26(m,6h),2.12(s,3h),1.26(d,j=3.6hz,2h).13cnmr(100mhz,cdcl3),如图10所示,δ202.52,170.42,147.94,139.12,129.91,128.90,128.50,125.84,118.34,110.39,69.78,64.89,61.82,58.78,34.56,29.35,27.04.hrms(esi)m/zcalculatedforc30h32n6o4[m+na]+:563.2398,found:563.2367.
实施例14
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3c即乙酰乙酸正戊酯(0.0450ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4f的产率为85%。
反应如下所示:
化合物4f:
1hnmr(400mhz,cdcl3),如图11所示,δ7.91(s,2h),7.82(d,j=7.3hz,4h),7.42(t,j=7.5hz,4h),7.34(t,j=8.5,6.2hz,2h),5.50(d,j=1.8hz,2h),5.15(d,j=1.7hz,2h),3.91(t,j=6.8hz,2h),3.61(d,j=15.8hz,2h),3.47(d,j=15.8hz,2h),2.12(s,3h),1.55(t,j=6.8hz,2h),1.35–1.13(m,6h),0.85(t,j=6.9hz,3h).13cnmr(100mhz,cdcl3),图12所示,δ202.44,147.93,139.17,129.85,128.86,128.47,125.80,118.17,109.98,76.81,66.57,61.70,34.43,27.88,27.77,26.95,22.18,13.89.hrms(esi)m/zcalculatedforc32h36n6o3[m+na]+:575.2698,found:575.2654.
实施例15
将化合物1a即苯乙炔(0.0370ml,0.25mmol)、化合物2即酚取代的烯基叠氮化合物(0.0683g,0.25mmol),化合物3d即正丁酰乙酸甲酯(0.0430ml,0.25mmol)、三乙胺(0.0700ml,2.5mmol),加入到1ml的乙腈中,在70℃的条件下溶解,随后向体系中加入碘化亚铜(0.0143g,0.075mmol)、四(三苯基膦)钯(0.0100g,0.005mmol),在氮气保护条件下,继续加热搅拌4小时。tlc检测底物消失,反应结束。将反应液冷却后倒入30ml水中,用二氯甲烷(3×10ml)萃取,合并有机相,无水硫酸镁干燥、抽滤,然后减压蒸馏除去有机溶剂,得到粘稠的液体,经过硅胶柱层析(洗脱液为v石油醚:v乙酸乙酯=2:1)得到化合物4g的产率为87%。
反应如下所示:
化合物4g:
1hnmr(400mhz,cdcl3),如图13所示,δ7.91(s,2h),7.81(d,j=7.4hz,4h),7.42(t,j=7.5hz,4h),7.35(t,j=8.5,6.1hz,2h),5.50(d,j=1.9hz,2h),5.13(d,j=1.8hz,2h),3.69–3.45(m,7h),2.44(t,j=7.3hz,2h),1.43(d,j=7.4hz,2h),1.26(d,j=3.8hz,1h),0.84(t,j=7.4hz,3h).13cnmr(100mhz,cdcl3),图14所示,δ204.77,170.97,147.91,139.26,129.92,128.93,128.90,128.49,125.85,118.21,109.94,61.42,52.94,41.11,34.55,16.94,13.52.hrms(esi)m/zcalculatedforc30h32n6o3[m+na]+:547.2398,found:547.2385.
实施例16
本实施例提供化合物4a、4b、4c、4d、4e、4f、4g抗炎效果测试,包括如下步骤:
1.取肝癌hepg2细胞以6×104/ml的细胞密度接种于培养板中,每孔100μl,置37℃、5%co2及饱和湿度的条件下培养过夜。
2.待其贴壁后,分别加入化合物4a、4b、4c、4d、4e、4f、4g使其终浓度达到1μg/ml、2μg/ml、3μg/ml、4μg/ml、5μg/ml,每组设5个复孔,每孔终体积为200μl。对照组加入等量的dmem培养液。
3.培养24h和48h后,每孔加入mtt(5mg/ml))20μl,继续培养4h。
4.离心去上清,每孔加入dmso150μl溶解结晶颗粒。
5.酶标仪于570nm波长测定吸光度(d),计算不同时间、不同浓度的阿霉素对hepg2细胞的增殖抑制率,实验重复3次。
6.计算增殖抑制率=[(对照组d570—实验组d570)/对照组d570]×100%。
ic50是指在用药后存活的细胞数量减少一半时所需的药物浓度。在mtt法中,就是以对照组吸光度od值减少一半所需的药物浓度为ic50。除此以外,半数抑制浓度的含义还相当于药物对培养细胞的最小致死剂量的平均值,作为反映药效的定量指标,在各种药物的筛选中广泛应用。
具体的,根据公式:抑制率=1-加药组od值/对照组od值,计算化合物的ic50值
经测试,所有化合物的ic50值均低于4.75μg/kg,
通过上述测试结果可以看出,本公开实施例中所有化合物均有抗炎效果。
以上所揭露的仅为本公开的优选实施例而已,当然不能以此来限定本公开之权利范围,因此依本公开申请专利范围所作的等同变化,仍属本公开所涵盖的范围。
- 还没有人留言评论。精彩留言会获得点赞!