一种芳香乙烯类衍生物的晶型及其制备方法和应用与流程
1.本发明涉及一种芳香乙烯类衍生物的晶型及其制备方法和应用。
背景技术:
2.人程序性死亡因子配体1(pd
‑
l1),又称b7
‑
h1,属于b7家族成员,广泛分布于外周组织和造血细胞。pd
‑
l1q全长cdna 870bp,编码一段含290个氨基酸的i型过膜蛋白,pd
‑
l1主要表达于成熟的cd4t细胞、cd8t细胞、b细胞、单核细胞、树突状细胞(dendritic cells,dcs)、巨噬细胞等造血细胞及一些非造血细胞,如内皮细胞、胰岛细胞、肥大细胞等的膜表面。其中pd
‑
l1在多种肿瘤中高表达,如肺癌、胃癌、多发性骨髓、黑色素瘤和乳腺癌等。程序性死亡因子1(programmed death
‑
1,pd
‑
1)是pd
‑
l1的主要受体,主要分布于t细胞、b细胞和nk细胞等免疫相关细胞,在自身免疫病、肿瘤、感染、器官移植、过敏、免疫特权等等的免疫应答过程中都扮演着重要角色。
3.pd
‑
l1通过与其受体程序性死亡因子1相互作用抑制t细胞的激活或诱导成熟t细胞凋亡,使得免疫反应受到抑制。肿瘤发展过程中,癌细胞会通过上调pd
‑
l1表达,诱导t细胞的凋亡,避免免疫系统对其的清除。pd
‑
l1靶向抗体药物能够通过特异性阻断pd
‑
1/pd
‑
l1的相互作用打破肿瘤免疫耐受,恢复肿瘤特异性t细胞对肿瘤细胞的杀伤功能,进而实现肿瘤的清除。
4.pd
‑
1/pd
‑
l1发挥着负性免疫调节作用。pd
‑
1/pd
‑
l1信号可抑制t细胞活化和增殖,与此同时,细胞因子白细胞介素2(il2)、干扰素γ和il
‑
10的分泌也减少(eur.j.immunol.,2002,32(3),634
‑
643.)。另外,pd
‑
1/pd
‑
l1信号对b细胞免疫功能也类似于t细胞,当pd
‑
1与b细胞抗原受体发生交联后,pd
‑
1细胞质区与含有蛋白酪氨酸酶2结合位点的酪氨酸酶发生作用,最终阻滞b细胞的活化。免疫负性调节分子pd
‑
1/pd
‑
l1在肿瘤免疫逃逸中的作用越来越引起人们的重视。大量研究证实,肿瘤微环境中的肿瘤细胞表面pd
‑
l1增高,同时与活化的t细胞上的pd
‑
1结合,传递负性调控信号,导致肿瘤抗原特异性t细胞的凋亡或免疫无能,从而抑制免疫反应,进而促使肿瘤细胞的逃逸。
5.目前已经上市的pd
‑
1/pd
‑
l1的抗体抑制剂有bms的nivolumab(2014),merck的lambrolizumab(2014),君实生物的toripalimab,信达生物的sintilimab,罗氏的atezolizumab和阿斯利康的durvalumab。相较于生物大分子,小分子化合物能够穿过细胞膜作用于细胞内靶点,小分子化合物经过化学修饰后往往具有更好的生物利用度和依从性,有效避免消化肠道中酶类的分解失活。
6.目前尚未有使用方便口服有效的小分子pd
‑
1/pd
‑
l1抑制剂上市,专利cn109988144a公开了一种芳香乙烯类pd
‑
l1小分子化合物,化学结构式为它对pd
‑
1和pd
‑
l1的结合有很强的抑制作用,恢复
肿瘤特异性t细胞对肿瘤细胞的杀伤功能,能够有效缓解和治疗与pd
‑
1/pd
‑
l1有关的癌症等相关疾病。
7.除了对激酶有较好的抑制活性以外,作为药用活性成分的晶型结构往往影响到该药物的化学稳定性,结晶形式、制备方法及储存条件的不同有可能导致化合物的晶型结构的变化,有时还会伴随着产生其他形态的晶型。一般来说,无定型的药物产品没有规则的晶型结构,往往具有其他缺陷,比如产物稳定性较差,析晶较细,易结块,流动性差等,这些差异往往导致生产放大时的困难。同时晶型对药物在生产、加工、储存、运输时的稳定性以及治疗时的生物利用度都有至关重要的影响,而且,从获得一种商业上可行的生产方法的角度或者从生产含有活性化合物的药用组合物的角度出发,活性成分的化学稳定性、固态稳定性和储存期限均是非常重要的因素,因此提供一种具有所需性质的药物的合适形式对于药物生产,储存是十分重要的。
技术实现要素:
8.本发明提供了一种芳香乙烯类衍生物的晶型及其制备方法和应用。本发明的晶型具有良好稳定性、不易吸湿、容易制得、对药物的优化和开发具有重要的价值。
9.本发明提供了一种如式i所示的化合物的晶型a,其以2θ角表示的x
‑
射线粉末衍射图,在9.923
±
0.2
°
、10.883
±
0.2
°
和17.357
±
0.2
°
处具有特征峰;
10.或者,在3.979
±
0.2
°
、9.923
±
0.2
°
、10.883
±
0.2
°
、17.357
±
0.2
°
、18.607
±
0.2
°
和19.294
±
0.2
°
处具有特征峰;
11.或者,在3.979
±
0.2
°
、4.991
±
0.2
°
、9.923
±
0.2
°
、10.883
±
0.2
°
、14.251
±
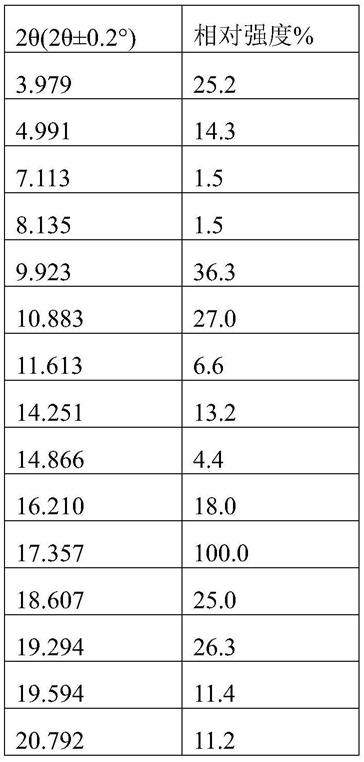
0.2
°
、16.210
±
0.2
°
、17.357
±
0.2
°
、18.607
±
0.2
°
、19.294
±
0.2
°
、19.594
±
0.2
°
和20.792
±
0.2
°
处具有特征峰;
12.或者,在3.979
±
0.2
°
、4.991
±
0.2
°
、7.113
±
0.2
°
、8.135
±
0.2
°
、9.923
±
0.2
°
、10.883
±
0.2
°
、11.613
±
0.2
°
、14.251
±
0.2
°
、14.866
±
0.2
°
、16.210
±
0.2
°
、17.357
±
0.2
°
、18.607
±
0.2
°
、19.294
±
0.2
°
、19.594
±
0.2
°
、20.792
±
0.2
°
、21.272
±
0.2
°
、24.437
±
0.2
°
、25.257
±
0.2
°
、26.2295
±
0.2
°
、27.870
±
0.2
°
、28.631
±
0.2
°
、29.126
±
0.2
°
、29.943
±
0.2
°
处具有特征峰;
[0013][0014]
在本发明某些优选实施方案中,所述的晶型a,其以2θ角表示的x
‑
射线粉末衍射图中,2θ值如表1所示;
[0015]
表1
[0016]
2θ(2θ
±
0.2
°
)相对强度%3.97925.24.99114.37.1131.5
8.1351.59.92336.310.88327.011.6136.614.25113.214.8664.416.21018.017.357100.018.60725.019.29426.319.59411.420.79211.221.2726.824.4376.725.2575.926.2294.427.8703.728.6311.929.1265.229.9431.5。
[0017]
在本发明某些优选实施方案中,所述的晶型a的偏光显微镜分析中,晶型形状优选为颗粒状或棒状,其粒径大小优选为10~100μm;所述的晶型a的偏光显微镜分析可在如下条件下进行:所述的显微镜优选为10倍物镜,所述的显微镜优选为正交偏光镜。所述的晶型a的偏光显微镜图优选如图1所示。
[0018]
在本发明某些优选实施方案中,所述的晶型a的差示扫描热分析中,在247℃有热吸收峰,其熔化热优选为118.0j/g;所述的晶型a的差示扫描热分析可在如下条件下进行:优选置于非密闭铝盘中进行,优选在氮气流(例如流量为50ml/min的氮气流)中进行,优选在25℃平衡,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型a的差示扫描量热分析图谱(dsc)优选如图2所示。
[0019]
在本发明某些优选实施方案中,所述的晶型a的热重分析中,样品从26.76℃至119.97℃,失重仅为0.1447%,所述“%”为重量百分比;所述的晶型a的热重分析可在如下条件下进行:优选置于铂金样品盘中进行,优选在氮气流(例如流量为60ml/min的氮气流)中进行,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型a的热重分析(tga)图谱优选如图3所示。
[0020]
在本发明某些优选实施方案中,所述的晶型a的x
‑
射线衍射分析中,所述的晶型a的x
‑
射线衍射分析可在如下条件下进行:优选在光源为cuk下进行,优选x
‑
射线强度为40kv/40ma,优选扫描模式为theta
‑
theta,优选角度范围为2
°
~40
°
(例如4
°
~40
°
),优选步长为0.05
°
,优选扫描速度为0.5秒/步。所述晶型a的x
‑
射线粉末衍射图图谱(xrpd)优选如
图4所示。
[0021]
在本发明某些优选实施方案中,所述的晶型a的动态水分吸附分析中,80%rh湿度时吸湿增重为0.310%,95%rh湿度时吸湿增重为0.409%,表明晶型a略有引湿性;所述的晶型a的动态水分吸附分析可在如下条件下进行:优选在温度为25℃下进行,优选在湿度0%rh条件下干燥60min后进行。所述的晶型a的动态水分吸附分析图谱(dvs)优选如图5所示。
[0022]
本发明提供了一种如式i所示的化合物的晶型b,其以2θ角表示的x
‑
射线粉末衍射图,在3.424
±
0.2
°
、6.576
±
0.2
°
和19.297
±
0.2
°
处具有特征峰;
[0023]
或者,在3.424
±
0.2
°
、6.576
±
0.2
°
、18.217
±
0.2
°
、19.297
±
0.2
°
、20.901
±
0.2
°
和26.379
±
0.2
°
处具有特征峰;
[0024]
或者,在3.424
±
0.2
°
、6.576
±
0.2
°
、14.467
±
0.2
°
、16.406
±
0.2
°
、17.567
±
0.2
°
、18.217
±
0.2
°
、19.297
±
0.2
°
、20.557
±
0.2
°
、20.901
±
0.2
°
、22.460
±
0.2
°
、25.084
±
0.2
°
、25.878
±
0.2
°
、26.379
±
0.2
°
和28.983
±
0.2
°
处具有特征峰;
[0025]
或者,在3.424
±
0.2
°
、6.576
±
0.2
°
、9.732
±
0.2
°
、11.304
±
0.2
°
、12.905
±
0.2
°
、13.918
±
0.2
°
、14.467
±
0.2
°
、16.406
±
0.2
°
、17.567
±
0.2
°
、18.217
±
0.2
°
、19.297
±
0.2
°
、20.557
±
0.2
°
、20.901
±
0.2
°
、22.460
±
0.2
°
、23.872
±
0.2
°
、25.084
±
0.2
°
、25.878
±
0.2
°
、26.379
±
0.2
°
、28.983
±
0.2
°
、29.531
±
0.2
°
、30.459
±
0.2
°
、32.171
±
0.2
°
、34.297
±
0.2
°
、37.676
±
0.2
°
和38.902
±
0.2
°
处具有特征峰;
[0026][0027]
在本发明某些优选实施方案中,所述的晶型b,以2θ角表示的x
‑
射线粉末衍射图中,2θ值如表2所示;
[0028]
表2
[0029][0030][0031]
在本发明某些优选实施方案中,所述的晶型b的x
‑
射线衍射分析中,所述的晶型b的x
‑
射线衍射分析可在如下条件下进行:优选在光源为cuk下进行,优选x
‑
射线强度为40kv/40ma,优选扫描模式为theta
‑
theta,优选角度范围为2
°
~40
°
(例如4
°
~40
°
),优选步长为0.05
°
,优选扫描速度为0.5秒/步。所述的晶型b的x
‑
射线粉末衍射图图谱(xrpd)优选如图6所示。
[0032]
在本发明某些优选实施方案中,所述的晶型b的差示扫描热分析中,在243℃有热吸收峰,其熔化热优选为93.73j/g;所述的晶型b的差示扫描热分析可在如下条件下进行:优选置于非密闭铝盘中进行,优选在氮气流(例如流量为50ml/min的氮气流)中进行,优选在25℃平衡,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型b的差示扫描量热分析图谱(dsc)优选如图7所示。
[0033]
在本发明某些优选实施方案中,所述的晶型b的热重分析中,从25.3℃加热到92.5℃有5.2%的失重,所述“%”为重量百分比;所述的晶型b的热重分析可在如下条件下进行:优选置于铂金样品盘中进行,优选在氮气流(例如流量为60ml/min的氮气流)中进行,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型b的热重分析(tga)图谱优选如图8所示。
[0034]
在本发明某些优选实施方案中,所述的晶型b的动态水分吸附分析中,在从0%相对湿度(rh)到95%rh样品增重7.235%,表明晶型b具有引湿性;所述的晶型b的动态水分吸附分析可在如下条件下进行:优选在温度为25℃下进行,优选在湿度0%rh条件下干燥60min后进行。所述的晶型b的动态水分吸附分析图谱(dvs)优选如图9所示。
[0035]
本发明提供了一种如式i所示的化合物的晶型c,其以2θ角表示的x
‑
射线粉末衍射图,在6.250
±
0.2
°
、18.458
±
0.2
°
和19.302
±
0.2
°
处具有特征峰;
[0036]
或者,在6.250
±
0.2
°
、8.779
±
0.2
°
、13.720
±
0.2
°
、18.458
±
0.2
°
和19.302
±
0.2
°
处具有特征峰;
[0037]
或者,在6.250
±
0.2
°
、8.779
±
0.2
°
、12.635
±
0.2
°
、13.720
±
0.2
°
、16.525
±
0.2
°
、18.458
±
0.2
°
和19.302
±
0.2
°
处具有特征峰;
[0038]
或者,在6.250
±
0.2
°
、6.979
±
0.2
°
、8.779
±
0.2
°
、12.635
±
0.2
°
、13.720
±
0.2
°
、16.525
±
0.2
°
、18.458
±
0.2
°
、19.302
±
0.2
°
、20.852
±
0.2
°
、22.345
±
0.2
°
、24.772
±
0.2
°
、25.230
±
0.2
°
、27.285
±
0.2
°
处具有特征峰;
[0039]
或者,在6.250
±
0.2
°
、6.979
±
0.2
°
、8.779
±
0.2
°
、12.635
±
0.2
°
、13.720
±
0.2
°
、15.285
±
0.2
°
、16.525
±
0.2
°
、18.458
±
0.2
°
、19.302
±
0.2
°
、20.852
±
0.2
°
、22.345
±
0.2
°
、24.772
±
0.2
°
、25.230
±
0.2
°
、25.996
±
0.2
°
、27.285
±
0.2
°
、28.303
±
0.2
°
、28.829
±
0.2
°
、29.699
±
0.2
°
、30.703
±
0.2
°
、33.133
±
0.2
°
、34.655
±
0.2
°
、36.829
±
0.2
°
、37.967
±
0.2
°
处具有特征峰;
[0040][0041]
在本发明某些优选实施方案中,所述的晶型c,其以2θ角表示的x
‑
射线粉末衍射图中,2θ值如表3所示;
[0042]
表3
[0043][0044][0045]
在本发明某些优选实施方案中,所述的晶型c的x
‑
射线衍射分析中,所述晶型c的x
‑
射线粉末衍射图图谱(xrpd)优选如图10所示。
[0046]
在本发明某些优选实施方案中,所述的晶型c的差示扫描热分析中,在243℃有热吸收峰,熔化热优选为99.33j/g;所述的晶型c的差示扫描热分析可在如下条件下进行:优选置于非密闭铝盘中进行,优选在氮气流(例如流量为50ml/min的氮气流)中进行,优选在25℃平衡,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型c的差示
扫描量热分析图谱(dsc)优选如图11所示。
[0047]
在本发明某些优选实施方案中,所述的晶型c的热重分析中,从24.0℃加热到58.0℃有0.62%的失重,从58.0℃加热到162.3℃有2.5%的失重,所述“%”为重量百分比;所述的晶型c的热重分析可在如下条件下进行:优选置于铂金样品盘中进行,优选在氮气流(例如流量为60ml/min的氮气流)中进行,优选以10℃/min的升温速率升温,优选从25℃加热至300℃。所述的晶型c的热重分析(tga)图谱优选如图12所示。
[0048]
在本发明某些优选实施方案中,所述的晶型c的动态水分吸附分析中,从0%rh到95%rh样品增重4.767%,表明晶型c具有引湿性;所述的晶型c的动态水分吸附分析可在如下条件下进行:优选在温度为25℃下进行,优选在湿度0%rh条件下干燥60min后进行。所述的晶型c的动态水分吸附分析图谱(dvs)优选如图13所示。
[0049]
本发明中,所述的x
‑
射线粉末衍射中使用的射线为kα射线。
[0050]
本发明中,所述的x
‑
射线粉末衍射中使用的靶型为cu靶。
[0051]
本发明还提供了一种如式i所示的化合物晶型a的制备方法,包括如下步骤:溶剂中,将如式i所示的化合物进行结晶,即可;所述的结晶方法为混悬液平衡法、溶液加热
‑
缓慢降温法或反溶剂法;所述的溶剂为乙醇,所述的结晶方法为反溶剂法时,反溶剂为烷烃类溶剂。
[0052]
所述的晶型a的制备方法中,所述的结晶方法为反溶剂法时,所述的烷烃类溶剂可为本领域常规的烷烃类溶剂,所述的烷烃类溶剂优选为c1‑
10
烷烃类溶剂,更优选为正庚烷。
[0053]
所述的晶型a的制备方法中,所述的结晶的温度可为本领域常规的温度,所述的结晶温度优选为20℃
‑
60℃(例如室温(20℃
‑
25℃)或50℃)。
[0054]
所述的晶型a的制备方法中,所述的如式i所示的化合物与所述的溶剂的质量体积比可为本领域常规的比例,优选为5mg/ml
‑
20mg/ml(例如7.7mg/ml
‑
20mg/ml)。
[0055]
所述的晶型a的制备方法中,对所述的结晶的时间可不作特殊限制,能够析出晶体即可,所述的结晶时间优选为1h
‑
20d(例如1h
‑
2h、5h
‑
6h或10d
‑
20d)。
[0056]
所述的晶型a的制备方法中,所述的结晶方法为反溶剂法时,所述的反溶剂与所述的溶剂的质量比可为本领域常规的比例,优选为5:1
‑
8:1(例如6.5:1)。
[0057]
所述的晶型a的制备方法中,其优选包括以下步骤:将所述的溶剂与所述的如式i所示的化合物混合,超声,旋转,离心,即得目标晶型。所述的旋转优选在遮光下进行。所述的旋转优选在室温下进行。所述的旋转优选置于labquaker旋转器上进行。所述的旋转优选进行10d
‑
20d。所述的离心结束后,还优选包括干燥的操作。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为5mg/ml
‑
20mg/ml(例如7.7mg/ml
‑
20mg/ml)。
[0058]
所述的晶型a的制备方法中,其优选包括以下步骤:将所述的溶剂和所述的如式i所示的化合物混合,加热溶解,缓慢降温至室温,离心,即得目标晶型。所述的加热溶解的温度优选为50℃
‑
60℃。所述的加热溶解优选为水浴加热。所述的加热溶解的同时优选包含搅拌的操作,所述的搅拌的转速优选为200rpm。所述的加热溶解结束后,优选包含保温的操作,所述的保温时间优选为15min。所述的加热溶解结束后,优选还包含趁热过滤的操作,所述的过滤优选采用0.45μm滤膜过滤。所述的缓慢降温的速率优选为6℃/h。所述的离心结束后,还优选包括溶剂挥干的操作。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为5mg/ml
‑
20mg/ml(例如7.7mg/ml
‑
20mg/ml)。
[0059]
所述的晶型a的制备方法中,其优选包括以下步骤:将所述的溶剂和所述的如式i所示的化合物混合,加热溶解,滴加所述的反溶剂后,自然降温,离心,即得目标晶型。所述的加热溶解的温度优选为50℃
‑
60℃。所述的加热溶解的同时优选包含搅拌的操作,所述的搅拌的转速优选为200rpm。所述的加热溶解结束后,优选包含保温的操作,所述的保温时间优选为15min。所述的加热溶解结束后,还优选包含趁热过滤的操作,所述的过滤优选采用0.45μm滤膜过滤。所述的滴加所述的反溶剂的同时优选包含搅拌的操作。所述的反溶剂与所述的溶剂的体积比优选为5:1
‑
8:1(例如6.5:1)。所述的滴加所述的反溶剂结束后优选包含保温的操作,所述的保温时间优选为10min。所述的离心结束后,还优选包括溶剂挥干的操作。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为5mg/ml
‑
20mg/ml(例如7.7mg/ml
‑
20mg/ml)。
[0060]
本发明还提供了一种如式i所示的化合物晶型b的制备方法,其包括如下步骤:溶剂中,将如式i所示的化合物进行结晶,即可;所述的结晶方法为混悬液平衡法或反溶剂法;所述的结晶方法为混悬液平衡法时所述的溶剂为水,或乙醇和水;所述的结晶方法为反溶剂法时,所述的溶剂为乙醇或四氢呋喃,所述的反溶剂为水。
[0061]
所述的晶型b的制备方法中,所述的水可为蒸馏水、去离子水、纯净水、自来水和矿泉水中的一种或多种。
[0062]
所述的晶型b的制备方法中,所述的结晶的温度可为本领域常规的温度,所述的结晶温度优选为20℃
‑
60℃(例如室温(20℃
‑
25℃)或50℃)。
[0063]
所述的晶型b的制备方法中,所述的如式i所示的化合物与所述的溶剂的质量体积比可为本领域常规的比例,优选为5mg/ml
‑
40mg/ml(例如7.7mg/ml、11.1mg/ml、20mg/ml或33.3mg/ml)。
[0064]
所述的晶型b的制备方法中,所述的结晶方法为混悬液平衡法时,所述的溶剂为乙醇和水时,所述的乙醇和所述的水的体积比优选为1:3
‑
1:5(例如1:4)。
[0065]
所述的晶型b的制备方法中,所述的结晶方法为反溶剂法时,所述的反溶剂与所述的溶剂的体积比优选为1:1
‑
4:1(例如1:1或2.7:1)。
[0066]
所述的晶型b的制备方法中,对所述的结晶的时间可不作特殊限制,能够析出晶体即可,所述的结晶时间优选为1h
‑
20d(例如1h
‑
2h、1d或17d)。
[0067]
所述的晶型b的制备方法中,所述的结晶方法为混悬液平衡法时,其优选包括以下步骤:将所述的溶剂与所述的如式i所示的化合物混合,打浆,分离固体,即得目标晶型。所述的打浆优选在室温下进行。所述的打浆优选在搅拌下进行。所述的搅拌优选为磁力搅拌。所述的打浆优选进行1d
‑
17d。所述的分离固体优选为离心或过滤。所述的分离固体结束后,还优选包含干燥的操作,所述的干燥优选在40℃下进行,所述的干燥优选进行4h。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为5mg/ml
‑
40mg/ml(例如7.7mg/ml、11.1mg/ml、20mg/ml或33.3mg/ml)。
[0068]
所述的晶型b的制备方法中,所述的结晶方法为反溶剂法时,其优选包括以下步骤:将所述的溶剂与所述的如式i所示的化合物混合,加热溶解,滴加所述的反溶剂后,自然降温,离心,即得目标晶型。所述的加热溶解的温度优选为50℃
‑
60℃。所述的加热溶解的同时优选包含搅拌的操作,所述的搅拌的转速优选为200rpm。所述的加热溶解结束后,优选包含保温的操作,所述的保温时间优选为15min。所述的加热溶解结束后,优选包含趁热过滤
的操作,所述的过滤优选采用0.45μm滤膜过滤。所述的滴加所述的反溶剂优选在搅拌时加入。所述的滴加所述的反溶剂结束后优选包含保温的操作,所述的保温时间优选为10min。所述的离心结束后,还优选包括溶剂挥干的操作。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为5mg/ml
‑
40mg/ml(例如7.7mg/ml、11.1mg/ml、20mg/ml或33.3mg/ml)。
[0069]
本发明还提供了一种如式i所示的化合物晶型c的制备方法,包括如下步骤:溶剂中,将如式i所示的化合物以混悬液平衡法进行结晶,即可;所述的溶剂为异丙醇、n,n
‑
二甲基乙酰胺,或丙酮和水。
[0070]
所述的晶型c的制备方法中,所述的溶剂为丙酮和水时,所述的丙酮与所述的水的体积比优选为7:1
‑
10:1(例如8:1)。
[0071]
所述的晶型c的制备方法中,所述的水可为蒸馏水、去离子水、纯净水、自来水和矿泉水中的一种或多种。
[0072]
所述的晶型c的制备方法中,所述的结晶的温度可为本领域常规的温度,所述的结晶温度优选为室温。
[0073]
所述的晶型c的制备方法中,所述的如式i所示的化合物与所述的溶剂的质量体积比可为本领域常规的比例,所述的如式i所示的化合物与所述的溶剂的质量体积比优选为10mg/ml
‑
50mg/ml(例如20mg/ml、40mg/ml或44.4mg/ml)。
[0074]
所述的晶型c的制备方法中,对所述的结晶的时间可不作特殊限制,能够析出晶体即可,所述的结晶时间优选为1d
‑
20d(例如1d、7d、10d或20d)。
[0075]
所述的晶型c的制备方法中,其优选包括以下步骤:将所述的溶剂与所述的如式i所示的化合物混合,打浆或旋转,分离固体,即得目标晶型。所述的混合结束后,还优选包含超声的操作,所述的超声优选进行1min。所述的晶型c的制备方法包含打浆时,所述的打浆优选进行1d,所述的打浆优选在搅拌下进行,所述的搅拌优选为磁力搅拌。所述的晶型c的制备方法包含旋转时,所述的旋转优选进行10
‑
20d,所述的旋转优选置于labquaker旋转器上进行,所述的旋转优选在遮光下进行。所述的分离固体优选为离心或过滤。所述的分离固体结束后,还优选包含干燥的操作。所述的干燥优选在70℃下进行,所述的干燥优选进行4h。所述的如式i所示的化合物与所述的溶剂的质量体积比优选为10mg/ml
‑
50mg/ml(例如20mg/ml、40mg/ml或44.4mg/ml)。
[0076]
本发明还提供了一种所述的如式i所述的化合物的晶型在制备药物中的应用。该化合物在预防、缓解和/或治疗癌症引起的相关疾病的药物中的应用;所述癌症优选肺癌、食管癌、胃癌、大肠癌、肝癌、鼻咽癌、脑肿瘤、乳腺癌、宫颈癌、血癌和骨癌中的一种或多种。所述的药物优选包含治疗有效量的上述的如式i所述的化合物的晶型a、b或c。
[0077]
本文中所使用的术语“d”指天,术语“h”指小时,术语“min”指分钟。
[0078]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0079]
本发明所用试剂和原料均市售可得。
[0080]
本发明的积极进步效果在于:
[0081]
1、现有技术中并没有记载如式i所述的化合物的晶型,本技术首次发现它的多种新晶型。通过大量的实验与筛选,挑选出晶型a、晶型b和晶型c作为候选对象。
[0082]
2、本发明制备得到的晶型a、晶型b和晶型c具有良好稳定性、不易吸湿、容易制得,
能够避免药物开发或是生产过程中发生转晶的风险,避免生物利用度以及药效发生改变,可以开发成适合临床使用的剂型,具有很强的经济价值。
[0083]
3、本发明还提供了如式i所示的化合物的新晶型的制备方法,操作简便且重现性高,溶剂不易残留,对环境友好,适合不同规模化生产。
附图说明
[0084]
图1为如式i所示的化合物晶型a的偏振光显微镜图。
[0085]
图2为如式i所示的化合物晶型a的dsc图。
[0086]
图3为如式i所示的化合物晶型a的tga图。
[0087]
图4为如式i所示的化合物晶型a的xrpd图。
[0088]
图5为如式i所示的化合物晶型a的dvs图。
[0089]
图6为如式i所示的化合物晶型b的xrpd图。
[0090]
图7为如式i所示的化合物晶型b的dsc图。
[0091]
图8为如式i所示的化合物晶型b的tga图。
[0092]
图9为如式i所示的化合物晶型b的dvs图。
[0093]
图10为如式i所示的化合物晶型c的xrpd图。
[0094]
图11为如式i所示的化合物晶型c的dsc图。
[0095]
图12为如式i所示的化合物晶型c的tga图。
[0096]
图13为如式i所示的化合物晶型c的dvs图。
[0097]
图14为如式i所示的化合物晶型v的xrpd图。
[0098]
图15为如式i所示的化合物晶型v的dsc图。
[0099]
图16为如式i所示的化合物晶型v的tga图。
[0100]
图17为如式i所示的化合物晶型vii的xrpd图。
[0101]
图18为如式i所示的化合物晶型vii的dsc图。
[0102]
图19为如式i所示的化合物晶型vii的tga图。
[0103]
图20为如式i所示的化合物晶型viii的xrpd图。
[0104]
图21为如式i所示的化合物晶型viii的dsc图。
[0105]
图22为如式i所示的化合物晶型viii的tga图。
[0106]
图23为如式i所示的化合物晶型ix的xrpd图。
[0107]
图24为如式i所示的化合物晶型ix的dsc图。
[0108]
图25为如式i所示的化合物晶型ix的tga图。
具体实施方式
[0109]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0110]
下述晶型的制备方法中,加入溶剂的体积(ml)=样品质量
×
体积倍数(ml),例如实施例1制备方法一中的乙醇的体积为:0.02
×
50ml=1ml。
[0111]
无定型化合物i(如式i所示的化合物)的制备:
[0112]
在室温条件下,往化合物5
‑
a(化合物5
‑
a的制备参照专利cn109988144a中方法获得)和(s)
‑2‑
甲基丝氨酸(65mg,0.54mmol)的甲醇(10ml)和二氯甲烷(10ml)混合溶液中加入冰醋酸(32.8mg,0.54mmol),反应液在室温条件下搅拌1小时。然后,加入氰基硼氢化钠(85.8mg,1.36mmol)搅拌16小时。减压浓缩反应液,剩余物溶于乙酸乙酯(50ml)中,用水(20ml)和饱和食盐水(20ml)洗涤,有机相经无水硫酸钠干燥,减压浓缩,剩余物用二氯甲烷:甲醇=10:1的流动相经硅胶薄层层析制备板分离,伴有化合物i的硅胶用甲醇(20ml
×
3)洗涤,洗涤液减压浓缩得到无定型化合物i(24mg,产率:18.7%)。lc
‑
ms(esi):m/z=468[m
‑
h]
‑
[0113]1h nmr(500mhz,cd3od)δ:8.17(s,1h),7.81
‑
7.79(d,j=8.5hz,1h),7.68
‑
7.62(m,2h),7.57
‑
7.55(d,j=8.0hz,1h),7.46
‑
7.43(m,2h),7.39
‑
7.28(m,5h),7.20
‑
7.19(d,j=7.0hz,1h),4.36
‑
4.28(q,2h),4.03
‑
4.00(d,j=12.5hz,1h),3.86
‑
3.84(d,j=12.5hz,1h),2.33(s,3h),1.57(s,3h)ppm.
[0114]
实施例1:晶型a的制备方法:
[0115]
制备方法一:称取20mg如式i所示的化合物到4ml玻璃瓶中,向玻璃瓶中加入50倍体积乙醇(etoh),超声1分钟得到样品的悬浊液,将悬浊液样品小瓶包裹铝箔遮光后置于labquaker旋转器上,开始在室温下(约20~25℃)360度旋转平衡,分别在10天和20天取样,离心,干燥后用xrpd表征,表征结果为晶型a。
[0116]
制备方法二:分别称取约20mg如式i所示的化合物到10ml玻璃小瓶中,向玻璃小瓶中加入130倍体积乙醇(etoh),将样品置于磁力加热搅拌器上,水浴温度约50℃,转速为200rpm,加热促进样品溶解,保温15min,趁热将溶液用0.45μm滤膜过滤,续滤液转移到新的玻璃瓶中,并以6℃/h的速率缓慢降温至室温(20~25℃),将析出固体的溶剂体系离心后取出固体,待溶剂挥干后用xrpd表征,表征结果为晶型a。
[0117]
制备方法三(反溶剂法):称取约80mg式i化合物样品到20ml玻璃小瓶中,向小瓶中加入130倍体积的良溶剂乙醇(etoh),将样品瓶置于磁力加热搅拌器上,水浴温度约50℃,转速为200rpm,加热促进样品溶解,保温15min,趁热将溶液用0.45μm滤膜过滤,续滤液转移到新的小瓶中,搅拌状态下往各小瓶中缓慢滴加850倍的反溶剂正庚烷,保温10min后,自然降温,后将析出固体的溶剂体系离心后取出固体,待溶剂挥干后用xrpd表征,表征结果为晶型a。
[0118]
实施例2:晶型b的制备方法:
[0119]
制备方法一:称取如式i所示的化合物200mg到小瓶中,往小瓶中加入10倍乙醇和40倍水,室温条件下磁力搅拌打浆一天,将溶液离心,收集固体,40℃下干燥4小时,将干燥后的固体表征,表征结果为晶型b。
[0120]
制备方法二:称取100mg如式i所示的化合物到小玻璃瓶中,向玻璃瓶中加入30倍体积水,室温下磁力搅拌打浆17天,减压过滤,40℃下干燥4h,通过xrpd表征,表征结果为晶型b。
[0121]
制备方法三(反溶剂法):分别称取约80mg式i化合物样品到20ml玻璃小瓶中,按照下表,分别往小瓶中加入适量体积的良溶剂(具体体积见表4),将样品瓶置于磁力加热搅拌器上,水浴温度约50℃,转速为200rpm,保持水浴温度促进样品溶解,保温15min,趁热将各溶液用0.45μm滤膜过滤,续滤液转移到新的小瓶中,搅拌状态下依次往各小瓶中缓慢滴加
不同的反溶剂,保温10min后,自然降温,后将析出固体的溶剂体系离心后取出固体,待溶剂挥干后用xrpd表征,表征结果为晶型b。
[0122]
表4
[0123][0124][0125]
实施例3:晶型c的制备方法:
[0126]
制备方法一:称取如式i所示的化合物样品200mg,加入20倍丙酮和2.5倍水,室温条件下,磁力搅拌打浆1天,然后将溶液离心,收集固体,将收集得固体70℃下干燥4小时,将干燥后的固体用xrpd表征,表征结果为晶型c。
[0127]
制备方法二:200mg如式i所示的化合物+25倍异丙醇(ipa),室温下磁力搅拌打浆7天,减压过滤,固体70℃干燥4h,对固体样品进行xrpd表征,表征结果为晶型c。
[0128]
制备方法三:称取20mg如式i所示的化合物到4ml玻璃瓶中,向玻璃瓶中加入50倍体积n,n
‑
二甲基乙酰胺(dma),超声1分钟得到样品的悬浊液,将悬浊液样品小瓶包裹铝箔遮光后置于labquaker旋转器上,开始在室温下(约20~25℃)360度旋转平衡,分别在10天和20天取样,离心,干燥后用xrpd表征,表征结果为晶型c。
[0129]
通过x
‑
射线粉末衍射图谱(xrpd)、差示扫描量热分析(dsc)、热失重分析仪(tga)或动态水吸附仪(dvs)等对所示化合物的a,b,c晶型进行结构测定、晶型研究等。
[0130]
实施例4:晶型a偏振光显微镜表征(如图1)
[0131]
取少量如式i所示的化合物的晶型a样品,置于带刻度的载玻片上,加适量液体石蜡使之分散,盖上盖玻片,置于显微镜10倍物镜下观察颗粒形状、大小和晶型性质,使用正交偏光镜显示样品的双折射性质和晶癖,并用数码摄像头拍照。
[0132]
结果显示:该样品在偏振光显微镜下有明显的双折射现象,呈颗粒状和棒状,粒径大小为10~100μm。
[0133]
实施例5:晶型a差示扫描量热分析(dsc)(如图2)
[0134]
称取如式i所示的化合物的晶型a样品3.1820mg,置于非密闭铝盘中,氮气流(50ml/min)环境中,样品在25℃平衡,然后以10℃/min的升温速率从25℃加热至300℃,结果详见表5。
[0135]
表5
[0136]
样品起始温度(℃)最高温度(℃)峰面积(j/g)晶型a246.01246.77118.0
[0137]
实施例6:晶型a热重分析(tga)(如图3)
[0138]
称取如式i所示的化合物的晶型a样品15.3240mg,置于铂金样品盘中,样品氮气流
(60ml/min)环境中,以10℃/min的升温速率从25℃加热至300℃,样品从26.76℃至119.97℃,失重仅为0.1447%,说明样品几乎不含水或溶剂,结果详见表6。
[0139]
表6
[0140][0141]
实施例7:晶型a粉末x
‑
射线衍射分析(xrpd)(如图4)
[0142]
光源为cuk,x
‑
射线强度为40kv/40ma,扫描模式为theta
‑
theta,扫描角度范围为4
°
~40
°
,步长为0.05
°
,扫描速度为0.5秒/步,结果详见表7。
[0143]
表7
[0144]
[0145][0146]
实施例8:晶型a的动态水分吸附分析(dvs)(如图5)
[0147]
称取适量如式i所示的化合物的晶型a样品,温度25℃,湿度0%rh条件下干燥60min后,测试湿度从0%rh~95%rh变化时样品的吸湿特征,以及湿度从95%rh~0%rh变化时样品的去湿特征。湿度每步长变化为5%rh,平衡标准为10min内重量变化率小于0.01%/min,最长平衡时间为2h,dvs测定结果显示晶型a在25℃,80%rh湿度时吸湿增重为0.310%,95%rh湿度时吸湿增重为0.409%,表明该样品略有引湿性。
[0148]
实施例9:晶型b粉末x
‑
射线衍射分析(xrpd)(如图6)
[0149]
光源为cuk,x
‑
射线强度为40kv/40ma,扫描模式为theta
‑
theta,扫描角度范围为4
°
~40
°
,步长为0.05
°
,扫描速度为0.5秒/步,结果详见表8。
[0150]
表8
[0151]
[0152][0153]
实施例10:晶型b的差示扫描量热分析(dsc)(如图7)
[0154]
称取如式i所示的化合物的晶型b样品1.5330mg,置于非密闭铝盘中,氮气流(50ml/min)环境中,样品在25℃平衡,然后以10℃/min的升温速率从25℃加热至300℃,结果详见表9。
[0155]
表9
[0156][0157]
实施例11:晶型b的热重分析(tga)(如图8)
[0158]
称取如式i所示的化合物的晶型b样品1.7310mg,置于铂金样品盘中,样品氮气流(60ml/min)环境中,以10℃/min的升温速率从25℃加热至300℃,起始样品从25.3℃加热到92.5℃有5.2%的失重。
[0159]
实施例12:晶型b的动态水分吸附分析(dvs)(如图9)
[0160]
称取适量如式i所示的化合物的晶型b样品,温度25℃,湿度0%rh条件下干燥60min后,测试湿度从0%rh~95%rh变化时样品的吸湿特征,以及湿度从95%rh~0%rh变化时样品的去湿特征。湿度每步长变化为5%rh,平衡标准为5min内重量变化率小于0.01%/min,最长平衡时间为2小时,结果显示从0%rh到95%rh样品增重7.235%,表明该样品具有引湿性。
[0161]
实施例13:晶型c的粉末x
‑
射线衍射分析(xrpd)(如图10)
[0162]
光源为cuk,x
‑
射线强度为40kv/40ma,扫描模式为theta
‑
theta,扫描角度范围为4
°
~40
°
,步长为0.05
°
,扫描速度为0.5秒/步,结果详见表10。
[0163]
表10
[0164]
编号2θ(2θ
±
0.2
°
)相对强度%
16.25010026.97915.338.77938.6412.63521.3513.72032.6615.2853.1716.52524.1817.6965.5918.45873.01019.30259.01120.85215.41222.34519.11324.77214.31425.23011.51525.9964.11627.28513.21728.3034.11828.8293.71929.6992.32030.7035.62133.1333.12234.6551.92336.8292.42437.9673.1
[0165]
实施例14:晶型c的差示扫描量热分析(dsc)(如图11)
[0166]
称取如式i所示的化合物的晶型c样品1.550mg,置于非密闭铝盘中,氮气流(50ml/min)环境中,样品在25℃平衡,然后以10℃/min的升温速率从25℃加热至300℃,结果详见表11。
[0167]
表11
[0168][0169]
实施例15:晶型c的热重分析(tga)(如图12)
[0170]
称取如式i所示的化合物的晶型c样品5.3570mg,置于铂金样品盘中,样品氮气流(60ml/min)环境中,以10℃/min的升温速率从25℃加热至300℃,样品从24.0℃加热到58.0℃有0.62%的失重,从58.0℃加热到162.3℃有2.5%的失重。将样品加热至200℃,晶型c转化为晶型a,结果详见表12。
[0171]
表12
[0172][0173]
实施例16:晶型c的动态水分吸附分析(dvs)(如图13)
[0174]
称取适量如式i所示的化合物的晶型c样品,温度25℃,湿度0%rh条件下干燥60min后,测试湿度从0%rh~95%rh变化时样品的吸湿特征,以及湿度从95%rh~0%rh变化时样品的去湿特征。湿度每步长变化为5%rh,平衡标准为5min内重量变化率小于0.01%/min,最长平衡时间为2小时,结果显示从0%rh到95%rh样品增重4.767%,表明该样品具有引湿性。
[0175]
实施例17:晶型a、b、c的平衡溶解度和溶解速率比较
[0176]
为进一步研究晶型a、b、c在纯水和体内生理相关介质中的平衡溶解度和溶解速率方面是否存在差异,分别测试了4个晶型样品在水、模拟胃液(sgf)、模拟禁食状态肠液(fassif)和模拟进食状态肠液(fessif)中的平衡溶解度和溶解速率。分别称取约20mg样品a、b、c到4ml玻璃小瓶中,依次分别加入3ml介质,超声15s,放入37℃摇床中,转速为200rpm,分别在0.5h,2h和24h取约1ml混悬液,离心,转速12000rpm,时间5min,如有必要则将上清液稀释(稀释剂:甲醇/水=9/1)合适倍数后,或直接用hplc测定其浓度,计算各样品的溶解度数值。各时间点溶解度测试结果如表13所示:
[0177]
表13
[0178]
[0179][0180]
备注:sgf:模拟胃液;fassif:模拟禁食状态肠液;fessif:模拟进食状态肠液溶解度测试结果表明:在水、sgf、fassif和fessif中,3个晶型的溶解速率和溶解度无显著区别。
[0181]
实施例18:晶型a、b、c固态稳定性实验
[0182]
分别称取晶型a、b、c样品到20ml无色透明玻璃瓶中,将样品瓶分别放置于对应的影响因素和加速条件下(湿度条件下的样品瓶使用锡箔纸封口后扎孔放置,其他样品加盖并盖紧后放置),分别于放置1周和2周后取出,采用hplc检测各种条件下的样品含量和有关物质;按上述同样的方法分别称取一份样品并放置于对应的影响因素和加速条件下,放置2周后取出,用于观察各样品外观和xrpd表征,考察各晶型的物理稳定性,测试方法详见表14和表15;同时分别精密称取如式i所示的化合物的晶型a、b、c样品各两份到20ml无色透明玻璃瓶中并加盖盖紧,放置于
‑
20℃冰箱保存,2周时取出作为hplc分析的标准样品,测试结果详见表16和表17。
[0183]
表14 固体稳定性考察的含量液相色谱方法
[0184]
[0185][0186]
表15 固体稳定性来考察的有关物质液相色谱方法
[0187][0188]
表16 晶型a、b、c固态稳定性结果
‑
含量和有关物质
[0189]
[0190][0191]
表17 晶型a、b、c的固态稳定性测试结果
‑
外观和晶型
[0192][0193]
结论:
[0194]
在高温、高湿、光照和加速四个条件下放置2周的晶型a、b、c的样品外观和初始样品一样,均为类白色;晶型a和晶型c与初始样品比较无显著性差异,物理稳定性良好,而晶型b在高温(60℃)条件下出现转晶,物理稳定性较差。
[0195]
晶型a、b、c受光照影响明显,2周时总有关物质分别增加11.18%、1.28%和1.56%,说明光照对晶型a、b、c影响显著,需要注意避光保存。
[0196]
对比例1:晶型v的制备(混悬液平衡法)
[0197]
称取式i化合物样品100mg到小瓶中,往小瓶中加入20倍甲醇,室温条件下磁力搅拌打浆14天,将溶液离心,收集固体,40℃下干燥4小时,将干燥后的固体表征,将此晶型定义为晶型v,晶型v的xrpd图如图14,晶型v的dsc图如图15,将晶型v加热至85℃,晶型v转为晶型a。
[0198]
图16为晶型v的tga图(如图16,样品从24.8℃加热到82.7℃有3.690%的失重)。
[0199]
对比例2:晶型vii的制备(溶液加热
‑
缓慢降温法)
[0200]
称取式i化合物样品100mg到玻璃小瓶中,往小瓶中加入100倍体积乙酸乙酯(ea)和20倍体积甲醇(meoh),将样品置于磁力加热搅拌器上,水浴温度50℃下磁力搅拌溶清,过滤,滤液以6℃/h的速率缓慢降温至室温,过夜,减压过滤,固体70℃下干燥4h,将干燥后的固体表征,将此晶型定义为晶型vii,晶型vii的xrpd图如图17,将样品加热至200℃,冷却到室温表征xrpd,结果显示晶型vii转为晶型a。
[0201]
图18为晶型vii的dsc图(如图18,dsc热流曲线显示其初熔点在241.9℃,在200℃前有多个吸放热峰)。
[0202]
图19为晶型vii的tga图(如图19,样品从26.2℃到112℃有0.75%的失重,从112℃到200℃有1.69%的失重)。
[0203]
对比例3:晶型viii的制备(混悬液平衡法)
[0204]
称取式i化合物样品100mg到小瓶中,往小瓶中加入30倍乙腈(acn)为样品质量(g)
×
体积倍数),室温条件下磁力搅拌打浆2天,减压过滤,收集固体40℃下干燥4小时,将干燥后的固体表征,将此晶型定义为晶型viii,晶型viii的xrpd图如图20。将样品加热至180℃,晶型viii样品转为晶型a。
[0205]
图21为晶型viii的dsc图(如图21,dsc热流曲线显示其初熔点在239.0℃,而100℃之前有一较宽的吸热峰)。
[0206]
图22为晶型viii的tga图(如图22,样品从26.1℃到85.2℃有3.409%的失重)。
[0207]
对比例4:晶型ix的制备(溶液加热
‑
缓慢降温法)
[0208]
称取式i化合物样品100mg到玻璃小瓶中,往小瓶中加入30倍体积n,n
‑
二甲基乙酰胺(dma),将样品置于磁力加热搅拌器上,水浴温度50℃下磁力搅拌溶清,过滤,滤液以6℃/h的速率缓慢降温至室温,减压过滤,固体70℃下干燥4h,将干燥后的固体表征,将此晶型定义为ix,晶型ix的xrpd图如图23。将样品加热至180℃,晶型ix样品转为晶型a。
[0209]
图24为晶型ix的dsc图(如图24,dsc热流曲线显示其初熔点为241.8℃,200℃之前有多个吸放热峰)。
[0210]
图25为晶型ix的tga图(如图25,样品从27.4℃到95℃仅有0.035%的失重,从95℃到200℃有0.841%的失重)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1