一种氟啶虫酰胺的合成方法与流程
1.本发明涉及有机合成技术领域,尤其涉及一种氟啶虫酰胺的合成方法。
背景技术:
2.氟啶虫酰胺2003年上市,以独特的机理作用,对蚜虫表现出极高的生物活性,是目前防治蚜虫方面的特效药,且与目前市售其他杀虫剂无交互抗性,加之对蜜蜂友善的特性使其成为市场竞逐的产品,有望在未来逐渐抢占新烟碱类、氟啶虫胺腈及氟吡呋喃酮等的市场份额。
3.根据可查阅到的专利文献显示,目前由4-三氟甲基烟酸或4-三氟甲基烟酸盐制备氟啶虫酰胺的方法大致可以归类直接法和间接法:直接法的代表是以4-三氟甲基烟酸和氯化亚砜、氨基乙腈硫酸盐为原料,以三乙胺为缚酸剂制备氟啶虫酰胺的方法,如:jp2007210923a,由于4-三氟甲基烟酰氯活性过高,生成氟啶虫酰胺的同时会继续与4-三氟甲基烟酰氯反应,得到n-氰甲基双(三氟甲基)烟酰胺这个副产物;n-氰甲基双(三氟甲基)烟酰胺可转化为氟啶虫酰胺,如cn107417606a;以4-三氟甲基烟酸、氨基乙腈盐酸盐和光气为原料,如:cn103951616a。间接法的代表以4-三氟甲基烟酸和1,3,5-三氰甲基六氢均三嗪为原料先制得n-氯甲基-n-氰甲基-4-(三氟甲基)烟酰胺,然后在盐酸作用下制得n-羟甲基-n-氰甲基-4-(三氟甲基)烟酰胺,最后在碳酸钠作用下制得氟啶虫酰胺的方法。但是不管是直接法还是间接法,都存在收率偏低,生产成本较高,不利于广泛的推广应用等的缺陷。进一步,有以甲基磺酰氯作为活化试剂,制备氟啶虫酰胺,既避免了制备高活性的4-三氟甲基烟酰氯,又能一步高效制得氟啶虫酰胺,如cn109851552a。虽然这个方法会产生固废且成本过高,但是指明了一个方向,寻找合适的试剂制备中间体,活性相对4-三氟甲基烟酰氯低,就可能可以高效经济生产氟啶虫酰胺。
4.因此,对氟啶虫酰胺的合成方法进行深入的研究,得出了本方法。
技术实现要素:
5.为了解决上述技术问题,本发明提供一种氟啶虫酰胺的合成方法,该方法能够有效提高氟啶虫酰胺的收率和纯度,并且该方法简便、安全和环保,适合大规模工业化生产。
6.为了实现上述目的,本发明采用技术方案为:
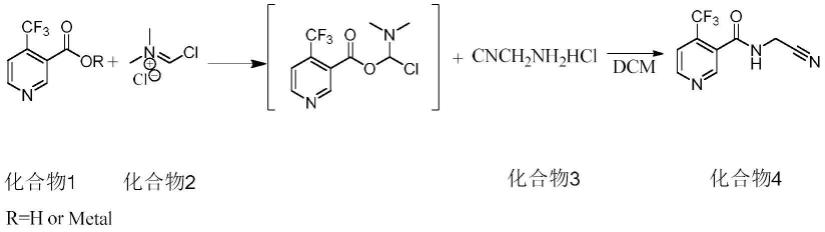
7.一种氟啶虫酰胺的合成方法,以4-三氟甲基烟酸或其金属盐为起始原料,生成式(i)或式(ii)所示结构的中间体,所述中间体与氨基乙腈盐进行取代反应,即得到所述氟啶虫酰胺;
[0008][0009]
其中,4-三氟甲基烟酸金属盐指4-三氟甲基烟酸的钠盐、钾盐、镁盐等,优选钠盐、
钾盐。
[0010]
发明人经过大量的试验发现,上述式(i)或式(ii)所示结构化合物,相比较4-三氟甲基烟酰氯活性较低,在与氨基乙腈盐进行取代反应时,反应选择性好,副产物少,目标产物收率高。
[0011]
具体地,所述氟啶虫酰胺的合成方法包括:以4-三氟甲基烟酸或其金属盐和vilsmeier试剂为原料,经加成反应,生成式(i)所示化合物。
[0012]
进一步地,所述vilsmeier试剂通过n,n-二取代的酰胺和酰氯经氯代反应制成。作为优选,所述n,n-二取代的酰胺为n,n-二甲基甲酰胺;所述酰氯为pocl3、氯化亚砜、草酰氯或邻苯二甲酰氯,优选氯化亚砜。
[0013]
进一步地,所述4-三氟甲基烟酸或其金属盐、氨基乙腈盐与vilsmeier试剂的摩尔比为1:(1~2):(1~3),优选1:1.1:1.1。
[0014]
进一步地,所述加成反应的温度为-80~50℃,优选-20~0℃;
[0015]
进一步地,所述取代反应的温度为-20~150℃,优选0~25℃。
[0016]
所述氟啶虫酰胺的另一合成方法包括:以4-三氟甲基烟酸或其金属盐和酰氯化试剂为原料,经取代反应,生成的酰氯产物与4-二甲氨基吡啶经亲电取代反应,生成式(ii)所示化合物。
[0017]
进一步地,所述酰氯化试剂为氯化亚砜、草酰氯、三氯化磷、五氯化磷或光气,优选氯化亚砜。
[0018]
进一步地,4-三氟甲基烟酸或其金属盐、酰氯化试剂、4-二甲氨基吡啶与氨基乙腈盐的摩尔比为1:(1~2):(1~2):(1~2),优选1:1.1:1.1:1.1。
[0019]
进一步地,所述方法在-10~150℃的温度下进行,优选0~25℃。
[0020]
在上述两种氟啶虫酰胺的合成方法中,所述方法在溶剂中进行,所述溶剂为二氯甲烷、二氯乙烷、二氧六环、甲基叔丁基醚、四氢呋喃甲苯、乙酸乙酯、乙腈中的一种或多种,优选二氯甲烷;
[0021]
进一步地,所述溶剂的重量为4-三氟甲基烟酸或其金属盐重量的3~8倍,优选3~5倍。
[0022]
进一步地,所述方法在碱存在或不存在的条件下进行。作为优选,所述碱选自吡啶、哌啶、三乙胺、4-二甲氨基吡啶、碳酸钠、碳酸钾中的一种或多种,进一步优选吡啶或不添加碱。
[0023]
进一步地,所述氨基乙腈盐为氨基乙腈盐酸盐或氨基乙腈硫酸盐,优选氨基乙腈盐酸盐。
[0024]
在一些具体的实施例中,所述氟啶虫酰胺的合成路线如下:
[0025]
[0026]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷,4-三氟甲基烟酸(化合物1,r=h)搅拌下降至0℃,缓慢滴加vilsmeier试剂(化合物2),保温1小时,添加氨基乙腈盐酸盐(化合物3),升至室温,保温2-3小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶得到产品(化合物4)。
[0027]
在另一些具体的实施例中,所述氟啶虫酰胺的合成路线如下:
[0028][0029]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷,4-三氟甲基烟酸(化合物1,r=h)搅拌下降至0℃,缓慢滴加氯化亚砜(化合物5),保温1小时,添加4-二甲氨基吡啶(化合物6),保温1小时,添加氨基乙腈盐酸盐(化合物3),升至室温,保温2-3小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶重结晶得到产品(化合物4)。
[0030]
在本发明的方法中,对反应的时间没有特别的限定,本领域技术人员可以通过采用常规方法例如gc、hplc进行检测分析,优选在起始原料4-三氟甲基烟酸或其金属盐的转化率达到99.0%以上时停止反应。
[0031]
本发明有益效果如下:
[0032]
(1)所得产品氟啶虫酰胺的收率和纯度均较高;
[0033]
(2)所用的原料对人体更加安全;
[0034]
(3)整个工艺更加安全、简便且成本低;
[0035]
(4)部分反应物料能够回收套用,进一步降低成本,并且更加环保。
[0036]
本发明通过以下两个新方法:第一,以4-三氟甲基烟酸或4-三氟甲基烟酸盐、vilsmeier试剂和氨基乙腈盐酸盐为原料;第二,以4-三氟甲基烟酸或4-三氟甲基烟酸盐、氯化亚砜、4-二甲氨基吡啶和氨基乙腈盐酸盐为原料,二氯甲烷为溶剂,能够在室温下安全、经济、高效、可广泛工业化制备氟啶虫酰胺。
具体实施方式
[0037]
以下实施例用于说明本发明,但不用来限制本发明的范围。实施例中未注明具体技术或条件者,按照本领域内的文献所描述的技术或条件,或者按照产品说明书进行。所用试剂或仪器未注明生产厂商者,均为可通过正规渠道商购买得到的常规产品。在没有特别说明的情况下,所用原料均采用市售产品。
[0038]
vilsmeier试剂的制备:在带有温度计和搅拌桨的反应瓶内,加入n,n-二甲基甲酰胺,降温至5-10℃,缓慢滴加当量的氯化亚砜,升温至50℃反应1-2小时,得到vilsmeier试剂,备用。
[0039]
实施例1
[0040]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.54g,搅拌下降至0℃,缓慢滴加69.86g vilsmeier试剂,保温1小时,添加氨基乙腈盐酸盐
50.90g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶得到115.39g,纯度95.54%,收率96.21%。
[0041]
实施例2
[0042]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.57g,搅拌下降至0℃,缓慢滴加69.88g vilsmeier试剂,保温1小时,添加氨基乙腈盐酸盐50.89g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶得到115.26g,纯度96.27%,收率96.81%。
[0043]
实施例3
[0044]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.58g,搅拌下降至0℃,缓慢滴加69.83g vilsmeier试剂,保温1小时,添加氨基乙腈盐酸盐50.87g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶得到113.57g,定量96.13%,收率95.24%。
[0045]
实施例4
[0046]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.55g,搅拌下降至0℃,缓慢滴加氯化亚砜65.40g,保温1小时,加入4-二甲氨基吡啶67.17g,保温1小时,添加氨基乙腈盐酸盐50.89g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶重结晶得到116.70g,纯度96.54%,收率98.31%。
[0047]
实施例5
[0048]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.59g,搅拌下降至0℃,缓慢滴加氯化亚砜65.42g,保温1小时,加入4-二甲氨基吡啶67.19g,保温1小时,添加氨基乙腈盐酸盐50.91g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶重结晶得到118.97g,纯度95.27%,收率98.87%。
[0049]
实施例6
[0050]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.57g,搅拌下降至0℃,缓慢滴加氯化亚砜65.41g,保温1小时,加入4-二甲氨基吡啶67.18g,保温1小时,添加氨基乙腈盐酸盐50.92g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶重结晶得到116.56g,纯度96.73%,收率98.37%。
[0051]
对比例
[0052]
在带有温度计,搅拌桨的反应瓶内,加入二氯甲烷450g,4-三氟甲基烟酸95.51g,搅拌下降至0℃,缓慢滴加氯化亚砜65.44g,保温1小时,添加氨基乙腈盐酸盐50.92g,升至室温,保温2小时,取样中控反应完成后,加入适量的氯化铵水溶液洗涤一次,有机相脱溶得到111.85g,纯度91.54%,收率89.42%。
[0053]
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1