一种恩格列净的合成方法
本发明属于原料药制备技术领域,涉及一种sglt-2抑制剂恩格列净的合成方法。
背景技术:
恩格列净(empagliflozin),于2014年8月由美国fda批准上市,是由勃林格殷格翰公司和礼来公司共同研发的抗糖尿病药物。该药物的作用靶点为钠葡萄糖共转运体2(sglt-2),即通过减少肾脏对葡萄糖的重吸收,降低肾糖阈,促进葡萄糖从尿液排出,从而降低血糖水平。该药主要用于治疗患有2型糖尿病的成年人。恩格列净属于吡喃葡萄糖基取代的苯基衍生物类化合物,化学名称为(1s)-1,5-脱水-1-c-[4-氯-3-[[4-[[(3s)-四氢-3-呋喃基]氧基]苯基]甲基]苯基]-d-葡萄糖醇。cas号864070-44-0,相对分子质量:450.91,分子式:c23h27clo7,其结构式为:
恩格列净现有合成方法:
第一种方法为原研公司勃林格殷格翰公司开发的方法:以2-氯-5-溴苯甲酸为起始原料,经酰氯化、付克酰基化、羰基还原、酸性水解得到苯酚衍生物,苯酚衍生物中酚羟基用叔丁基二甲基氯硅烷保护后,在正丁基锂作用下,与糖发生亲核加成,接着甲醚化,用et3sih/bf3.et2o体系还原消除端基碳上的羟基,再与(r)-3-对甲苯磺酸四氢呋喃酯发生亲核取代反应,得到最终产品恩格列净。该工艺路线较长,总收率仅为12%。
方法二是latli等人报道了一种制备恩格列净的新方法(j.labelcompd.radiopharm2014,57,687-694):先构建三元环体系,再经正丁基锂拔溴后与糖亲核加成,总收率13.6%。
分析两条路线,合成恩格列净过程中最为重要的即是与糖亲核加成形成碳苷的过程,该步反应条件苛刻,如需要零下78℃反应条件,催化剂为易燃物质正丁基锂,且反应时间为10小时左右,不利于工业化生产。有鉴于此,本发明致力开发一条试剂廉价易得、合成步骤短、操作简单,收率高,具有成本优势的新型合成路线。
技术实现要素:
本发明提供了一种新的恩格列净的合成方法,旨在解决现有技术制备恩格列净的合成方法起始原料不易获取、反应条件苛刻、工艺路线繁琐等问题。
本发明提供一种新的恩格列净的制备方法,其合成路线如下:
具体的,本发明所述方法包括如下步骤:
(1)将化合物2制备成活性中间体3后不经纯化,直接催化剂的作用下与氯苯反应得到中间体4;
其中,pg为乙酰基、苯甲酰基、特戊酰基或叔丁酰基;
(2)在相转移催化剂存在下,中间体4与(s)-3-苯氧基四氢呋喃和多聚甲醛反应,制备得到中间体5:
(3)在碱性条件下脱出中间体5的保护基得到终产物恩格列净:
作为优选,所述步骤(1)中间体2制备成中间体3,反应溶剂为二氯甲烷、乙腈、乙酸乙酯或丙酮中的任一种,傅酸剂为三乙胺、dmap或吡啶中的任一种,优选吡啶,反应温度在0℃~30℃,优选为0℃,中间体2与三氟乙酸酐的摩尔比为1:1-2,优选摩尔比为1:1。
作为优选,中间体3制备成中间体4,催化剂为三氟化硼/乙醚,反应温度在0℃~10℃,中间体3与氯苯的摩尔比为1:2-4,优选摩尔比为1:2。
作为优选,所述步骤(2)的相转移催化剂可为冠醚、四丁基溴化铵或四丁基氟化铵中的任一种,优选为四丁基溴化铵,反应温度为0~100℃,优选为60~80℃,中间体4与(s)-3-苯氧基四氢呋喃、多聚甲醛和相转移催化剂的摩尔比为1:1-2:1-2:2-4,优选摩尔比为1:1:1:2。
作为优选,所述步骤(3)中所述的碱为氢氧化钠、氢氧化钾、甲醇钠中的任一种,优选甲醇钠,中间体5与碱摩尔比为的2-4倍,优选摩尔比为1:2,反应溶剂为甲醇、丙酮或乙醇中的任一种,优选甲醇。
本发明还涉及关键中间体2的制备方法,采用如下的技术方案:
关键中间体2的制备方法,包括如下步骤:
(a)以葡萄糖为起始原料,在缚酸剂存在下与各种酰化试剂反应得到中间体1;
(b)中间体1与乙酸肼反应得到中间体2;
作为优选,所述步骤(a)的酰化试剂为乙酰氯、乙酸酐、苯甲酰氯、特戊酰氯或叔丁酰氯中的任一种,优选乙酸酐,缚酸剂为三乙胺、dmap或吡啶中的任一种,优选吡啶,反应溶剂为二氯甲烷、乙腈、四氢呋喃或乙酸乙酯中的任一种,优选二氯甲烷;反应温度为0℃~30℃,优选为0℃,葡萄糖与酰化试剂的摩尔比为1:4-6,优选摩尔比为1:5。
作为优选,所述步骤(b)的反应溶剂为n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、乙腈、四氢呋喃、1,4-二氧六环、甲苯、异丙醇或丙酮中的任一种,优选n,n-二甲基甲酰胺,反应温度为0~30℃,中间体1与乙酸肼的摩尔比为1:1-2,优选摩尔比为1:1。
本发明提供了一种新的恩格列净的合成方法,解决现有技术制备恩格列净的合成方法起始原料不易获取、反应条件苛刻、工艺路线繁琐等问题,本发明合成路线短,反应中所用物料均容易得到,反应操作简单。
附图说明
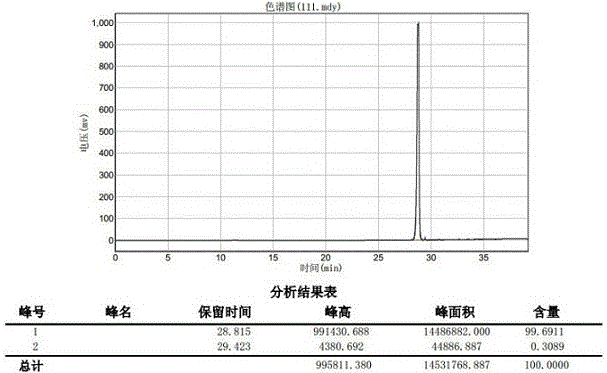
图1为恩格列净高效液相色谱图
具体实施方式
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
实施例1
中间体1的制备:
三颈瓶中加入葡萄糖(18g,100mmol)、二氯甲烷(100ml)和吡啶(10ml),冰水浴下滴加乙酸酐(47.2ml,500mmol),滴加完成后保持温度20~30℃反应3~4小时。冷却至0~5℃,加入水(100ml)搅拌10分钟后,混合液用二氯甲烷萃取3次,合并有机相,有机相用稀盐酸萃取1次,水洗2次,饱和碳酸氢钠洗1次,饱和食盐水洗1次,硫酸钠干燥。过滤浓缩后得化合物1(38.6g,收率99%)。
实施例2
中间体1的制备:
三颈瓶中加入葡萄糖(18g,100mmol)、二氯甲烷(100ml)和三乙胺(10ml),冰水浴下滴加乙酸酐(47.2ml,500mmol),滴加完成后保持温度20~30℃反应3~4小时。冷却至0~5℃,加入水(100ml)搅拌10分钟后,混合液用二氯甲烷萃取3次,合并有机相,有机相用稀盐酸萃取1次,水洗2次,饱和碳酸氢钠洗1次,饱和食盐水洗1次,硫酸钠干燥。过滤浓缩后得化合物1(35.1g,收率90%)。
实施例3
中间体1的制备:
三颈瓶中加入葡萄糖(18g,100mmol)、二氯甲烷(100ml)和吡啶(10ml),冰水浴下滴加乙酸酐(37.8ml,400mmol),滴加完成后保持温度20~30℃反应3~4小时。冷却至0~5℃,加入水(100ml)搅拌10分钟后,混合液用二氯甲烷萃取3次,合并有机相,有机相用稀盐酸萃取1次,水洗2次,饱和碳酸氢钠洗1次,饱和食盐水洗1次,硫酸钠干燥。过滤浓缩后得化合物1(32.0g,收率82%)。
实施例4
中间体1的制备:
三颈瓶中加入葡萄糖(18g,100mmol)、二氯甲烷(100ml)和吡啶(10ml),冰水浴下滴加乙酸酐(56.6ml,600mmol),滴加完成后保持温度20~30℃反应3~4小时。冷却至0~5℃,加入水(100ml)搅拌10分钟后,混合液用二氯甲烷萃取3次,合并有机相,有机相用稀盐酸萃取1次,水洗2次,饱和碳酸氢钠洗1次,饱和食盐水洗1次,硫酸钠干燥。过滤浓缩后得化合物1(38.2g,收率98%)。
实施例5
中间体2的制备:
三颈瓶中加入化合物1(39.0g,100mmol)、n,n-二甲基甲酰胺(60ml)和乙酸肼(7.37ml,100mmol),20~30℃下反应4~6小时。反应结束加入水(100ml),混合液用乙酸乙酯萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后用石油醚和乙酸乙酯混合溶剂重结晶得2(33.1g,95%)。
实施例6
中间体2的制备:
三颈瓶中加入化合物1(39.0g,100mmol)、乙腈(60ml)和乙酸肼(7.37ml,100mmol),20~30℃下反应4~6小时。反应结束加入水(100ml),混合液用乙酸乙酯萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后用石油醚和乙酸乙酯混合溶剂重结晶得2(29.6g,85%)。
实施例7
中间体2的制备:
三颈瓶中加入化合物1(39.0g,100mmol)、n,n-二甲基甲酰胺(60ml)和乙酸肼(14.7ml,200mmol),20~30℃下反应4~6小时。反应结束加入水(100ml),混合液用乙酸乙酯萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后用石油醚和乙酸乙酯混合溶剂重结晶得2(29.6g,85%)。
实施例8
中间体3的制备:
三颈瓶中加入中间体2(31.3g,90mmol)和二氯甲烷(100ml)和吡啶(10ml),搅拌溶解后冰水浴冷却至0~5℃,滴加三氟乙酸酐(12.5ml,90mmol),20~30℃下反应3~5小时。冰水浴条件下滴加50ml水淬灭反应,混合液用二氯甲烷萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后得化合物3(24.0g,60%)。
实施例9
中间体3的制备:
三颈瓶中加入中间体2(31.3g,90mmol)、二氯甲烷(100ml)和三乙胺(10ml),搅拌溶解后冰水浴冷却至0~5℃,滴加三氟乙酸酐(12.5ml,90mmol),20~30℃下反应3~5小时。冰水浴条件下滴加50ml水淬灭反应,混合液用二氯甲烷萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后得化合物3(20.0g,50%)。
实施例10
中间体3的制备:
三颈瓶中加入中间体2(31.3g,90mmol)、二氯甲烷(100ml)和吡啶(10ml),搅拌溶解后冰水浴冷却至0~5℃,滴加三氟乙酸酐(25.0ml,180mmol),20~30℃下反应3~5小时。冰水浴条件下滴加50ml水淬灭反应,混合液用二氯甲烷萃取3次,合并有机相水洗2次,饱和食盐水洗一次,硫酸钠干燥,过滤,浓缩后得化合物3(23.3g,56%)。
实施例11
中间体4的制备:
在三颈瓶中加入化合物3(22.2g,50mmol)、氯苯(10.2ml,100mmol)和乙腈(100ml),溶解后加入三氟化硼/乙醚(10ml),回流3~4小时。冰水浴滴加水(50ml)淬灭反应,反应液浓缩后用乙酸乙酯萃取3次,浓缩有机相,饱和氯化钠溶液洗两2次,无水硫酸钠干燥,浓缩后用乙酸乙酯石油醚重结晶分离得化合物4(11.0g,50%)。ms(esi)m/z=465.1[m+na]+;1hnmr(600mhz,dmso)δ7.65(d,j=7.1hz,2h),7.32(s,2h),6.35(d,j=6.9hz,1h),6.03(t,j=8.5hz,1h),5.88–5.81(m,2h),5.18(d,j=8.7hz,2h),4.64(d,j=8.5hz,1h),2.08(s,3h),2.06(s,3h),2.04(s,3h),2.02(s,3h).
实施例12
中间体4的制备:
在三颈瓶中加入化合物3(22.2g,50mmol)、氯苯(20.4ml,200mmol)和乙腈(100ml),溶解后加入三氟化硼/乙醚(10ml),回流3~4小时。冰水浴滴加水(50ml)淬灭反应,反应液浓缩后用乙酸乙酯萃取3次,浓缩有机相,饱和氯化钠溶液洗两2次,无水硫酸钠干燥,浓缩后用乙酸乙酯石油醚重结晶分离得化合物4(8.8g,40%)。
实施例13
中间体5的制备:
将化合物4(4.4g,10mmol)溶于乙腈中(100ml),溶解后加入四丁基溴化铵(3.2g,20mmol)、(s)-3-苯氧基四氢呋喃(1.6g,10mmol)和多聚甲醛(5.0g),反应温度升至80℃,反应10小时。反应完毕后,向反应液中加入稀盐酸(50ml),用二氯甲烷萃取3次,合并有机相,硫酸钠干燥,过滤,浓缩后用乙酸乙酯溶解,加入石油醚析出固体,得化合物5(4.0g,65%)。
实施例14
中间体5的制备:
将化合物4(4.4g,10mmol)溶于乙腈中(100ml),溶解后加入四丁基溴化铵(6.4g,40mmol)、(s)-3-苯氧基四氢呋喃(3.2g,20mmol)和多聚甲醛(10.0g),反应温度升至80℃,反应10小时。反应完毕后,向反应液中加入稀盐酸(50ml),用二氯甲烷萃取3次,合并有机相,硫酸钠干燥,过滤,浓缩后用乙酸乙酯溶解,加入石油醚析出固体,得化合物5(3.9g,63%)。
实施例15
中间体5的制备:
将化合物4(4.4g,10mmol)溶于乙腈中(100ml),溶解后加入四丁基氟化铵(5.2g,20mmol)、(s)-3-苯氧基四氢呋喃(1.6g,10mmol)和多聚甲醛(5.0g),反应温度升至80℃,反应10小时。反应完毕后,向反应液中加入稀盐酸(50ml),用二氯甲烷萃取3次,合并有机相,硫酸钠干燥,过滤,浓缩后用乙酸乙酯溶解,加入石油醚析出固体,得化合物5(3.7g,60%)。
实施例16
终产物恩格列净的制备:
在三颈瓶中加入化合物5(6.2g,10mmol)和甲醇(50ml),全部溶解后加入1mol/l氢氧化钾水溶液(20ml),室温反应10分钟。用阳离子交换树脂(h+型)调节反应液ph=7,将反应液浓缩后用甲苯和乙醇的混合溶剂重结晶得终产物恩格列净(4.5g,90%)。
实施例17
终产物恩格列净的制备:
在三颈瓶中加入化合物5(6.2g,10mmol)和甲醇(50ml),全部溶解后加入1mol/l的氢氧化钠水溶液(20ml),室温反应10分钟。用阳离子交换树脂(h+型)调节反应液ph=7,将反应液浓缩后用甲苯和乙醇的混合溶剂重结晶得终产物恩格列净(4.1g,88%)。
实施例18
终产物恩格列净的制备:
在三颈瓶中加入化合物5(6.2g,10mmol)和甲醇(50ml),全部溶解后加入1mol/l的甲醇/甲醇钠溶液(40ml),室温反应10分钟。用阳离子交换树脂(h+型)调节反应液ph=7,将反应液浓缩后用甲苯和乙醇的混合溶剂重结晶得终产物恩格列净(3.6g,80%)。
实施例19
终产物恩格列净的制备:
在三颈瓶中加入化合物5(6.2g,10mmol)和甲醇(50ml),全部溶解后加入1mol/l的甲醇/甲醇钠溶液(20ml),室温反应10分钟。用阳离子交换树脂(h+型)调节反应液ph=7,将反应液浓缩后用甲苯和乙醇的混合溶剂重结晶得终产物恩格列净(4.5g,99%),纯度为99.69%。
所得到的产物恩格列净经质谱和核磁共振确认结构,结果为:ms(esi)m/z=473.1[m+na]+;1hnmr(600mhz,dmso-d6)7.80(d,j=8.4hz,1h),7.35(d,j=2.0hz,1h),7.22(dd,j=2.0,8.4hz,1h),7.16(abq,j=8.8hz,2h),6.80(abq,j=8.8hz,1h),4.94(m,3h),4.82(d,j=6.4hz,1h),4.44(t,j=6.0hz,1h),3.96(m,3h),3.88-3.68(m,5h),3.46-3.42(m,1h),3.25-3.09(m,4h),2.24-2.16(m,1h),1.98-1.88(m,1h)。
- 还没有人留言评论。精彩留言会获得点赞!