一种吲哚类化合物及其制备方法与应用
本发明属于医药化学
技术领域:
,特别涉及一种吲哚类化合物及其制备方法与应用。
背景技术:
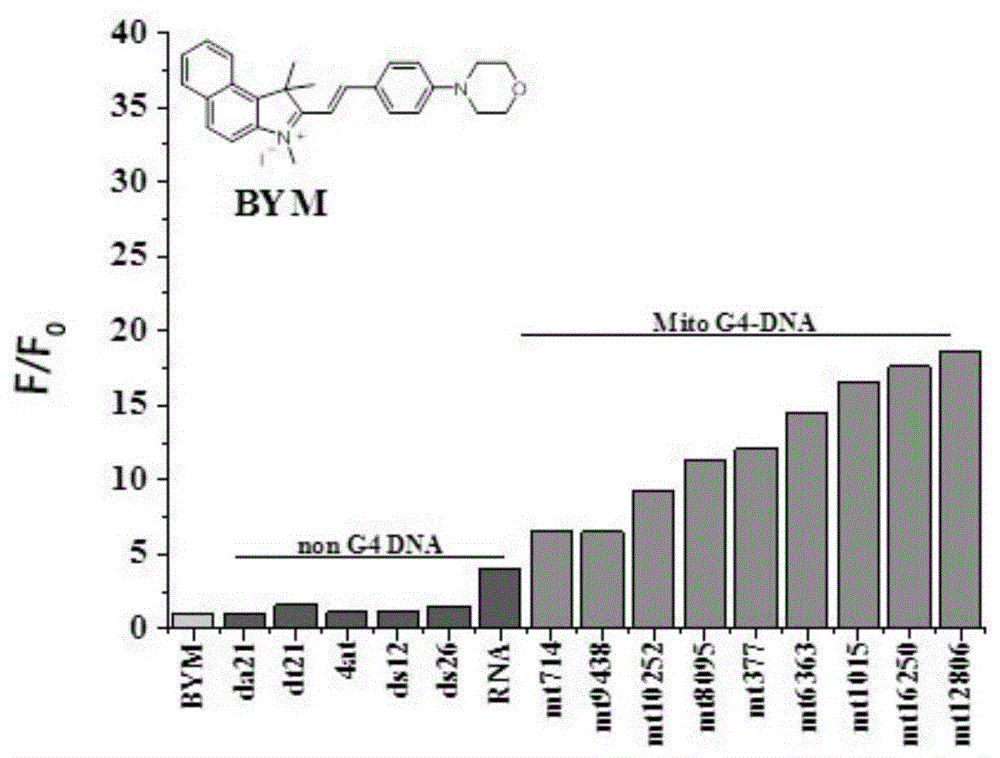
:线粒体是一种在能量代谢和细胞自我调节中起到重要作用的细胞器,其主要功能是将代谢物脱下的氢通过多种酶及辅酶所组成的传递体系传递,最后与氧结合生成水。由供氢体、传递体、受氢体以及相应的酶系统所组成的代谢系统,并且受氢体为氧,称为呼吸链。呼吸链由nad或nadh辅酶、黄素酶、铁硫蛋白、细胞色素等部分组成,其相互组合可以形成复合物i、ii、iii、iv、v,在呼吸链的电子传递中起着重要作用。癌细胞能量代谢旺盛,细胞内的线粒体数量相较于正常细胞增加。人类线粒体dna存在于线粒体基质中,16.5kb,负责编码线粒体16s和12s核糖体rna、22个线粒体trna和13个呼吸链蛋白。线粒体dna为环状双链,没有内含子,缺乏保护的组蛋白,外链富含鸟嘌呤,且称为重链,加上内部环境钾离子浓度高达150mm,易形成g-四链体;内链富含胞嘧啶,称为轻链。目前一些证据显示,线粒体g-四链体可能具有改变线粒体序列的功能,同时也被认为具有调控线粒体dna复制、转录和翻译等过程的潜在能力。目前已有许多g-四链体荧光配体被开发出来,但大多是靶向细胞核内的g-四链体的荧光配体,针对线粒体g-四链体开发的配体寥寥无几。因此,针对线粒体g-四链体的荧光配体的设计与研发具有重大意义,不仅能够实现对线粒体进行检测与诊断,而且可能开发出靶向线粒体的抗肿瘤药物。技术实现要素:本发明目的在于针对上述现有技术的不足之处而提供一种吲哚类化合物及其制备方法与应用。本发明通过对吲哚结构的改造,引入苯乙烯基和氮杂环得到所述吲哚类化合物,所述吲哚类化合物能够检测线粒体,具有发展成线粒体功能诊断试剂和抗肿瘤药物的潜力。为实现上述目的,本发明采用的技术方案如下:一种吲哚类化合物,其结构如式(i)所示:式(i)中,所述m为吲哚苯环上取代基r1的个数,m为0~4的自然数;m=2~4时,表示吲哚苯环上的多个h被取代基r1取代,不同取代位置上的取代基r1为相同基团或不同基团;r1为氢、取代苯环、芳香杂环、卤素、甲硫基、酯基、c1~c6的烷氧基、c2~c5的亚胺基、c1~c6的烷基或c1~c6的醇羟基;r2、r3分别独立选自c1~c6的烷氧基、c2~c5的亚胺基和c1~c6的烷基中的一种;r4为氢、甲基、羟基、酰胺基、羧基、磷酸基中的一种;r6为4-甲基哌嗪或吗啡啉;所述n为碳原子数,n=1~6的自然数。本发明所述吲哚类化合物是一种对线粒体g-四链体具有选择性结合的荧光配体。在体外实验中能够特异性识别线粒体g-四链体dna;在细胞实验中能够对细胞中的线粒体进行染色,并且具有荧光强度高、结合常数强,检测限低等优点,另外,本发明所述吲哚类化合物配体能够调控并影响线粒体功能,具有发展成线粒体功能诊断试剂和抗肿瘤药物的潜力。作为本发明的优选实施方式,所述m为吲哚苯环上取代基r1的个数,m为0~4的自然数;m=0时,吲哚苯环上的h不被取代;m=1时,表示吲哚苯环上的一个h被取代基r1;m=2-4时,表示吲哚苯环上的h被取代基r1取代的个数为2-4,不同取代位置上的取代基r1为相同基团或不同基团;r1独立选自氢、取代苯环、芳香杂环、卤素、甲硫基、酯基、c1~c6的烷氧基、c2~c5的亚胺基、c1~c6的烷基或c1~c6的羟基。作为本发明的优选实施方式,所述取代苯环的结构式为所述a为取代苯环上取代基r5的个数,a为0~5的自然数;a=2~5时,代表取代苯环上多个氢被多个取代基r5的个数,不同取代位置上的取代基r5为相同基团或不同基团;所述r5为-h、-f、-br、-no2、-n(ch3)2、-nh2、4-甲基哌嗪-1-基、哌嗪、吡啶、4-吗啡啉基、-oh、-och3中的一种。作为本发明的优选实施方式,所述a为取代苯环上取代基r5的个数,a为0~5的自然数;a=0时,取代苯环上的h不被取代;a=1时,表示取代苯环上的一个h被取代基r1;a=2-5时,表示取代苯环上的h被取代基r1取代的个数为2-5,不同取代位置上的取代基r5为相同基团或不同基团;所述r5独立选自-h、-f、-br、-no2、-n(ch3)2、-nh2、4-甲基哌嗪-1-基、哌嗪、吡啶、4-吗啡啉基、-oh、-och3中的一种。作为本发明的优选实施方式,所述芳香杂环为五元含氮杂环、六元含氮杂环、吲哚基或取代吲哚基。作为本发明的优选实施方式,所述吲哚类化合物的结构式为式(1)~(4)中的一种:本发明还要求保护所述吲哚类化合物的制备方法,包括如下步骤:(1)将反应物a加入溶剂a中混合均匀,再加入r4(ch2)nx,在60~70℃下反应24~30h,得到固体产物;(2)将步骤(1)中的固体产物和反应物b加入溶剂b中混合均匀,再加入催化剂,在60~100℃下反应12~24h,得到所述吲哚类化合物;所述反应物a的结构式为:所述反应物b的结构式为:所述r4(ch2)nx中,x为cl、br、i中的一种。本发明所述吲哚类化合物的合成路线如下:作为本发明的优选实施方式,所述溶剂b为乙醇、正丁醇、甲醇中的一种;所述催化剂为4-甲基哌啶、哌啶、吗啡啉、吡啶中的一种;所述溶剂a为环丁砜。作为本发明的优选实施方式,所述步骤(1)中,反应物a和r4(ch2)nx的摩尔比为1:1~5;反应物a的物质的量与溶剂a的体积配比为0.1~0.5mol/l。作为本发明的优选实施方式,所述步骤(2)中,固体产物和反应物b的摩尔比为1.5:1.8~3.0;反应物b的物质的量与溶剂b的体积配比为0.45~0.75mol/l;所述溶剂b与催化剂的体积比为4:0.1~0.3。作为本发明的优选实施方式,所述步骤(1)中,反应结束后加入乙酸乙酯静置沉淀,真空抽滤后用乙酸乙酯进行清洗,得到固体产物。作为本发明的优选实施方式,所述步骤(2)中,反应结束后有沉淀析出,将沉淀的各组分通过柱层析的方式分离,得到所述吲哚类化合物。作为本发明的优选实施方式,所述步骤(1)和步骤(2)均在防爆瓶中进行回流反应。作为本发明的优选实施方式,所述步骤(1)中,超声振荡使反应物a和溶剂a混合均匀。作为本发明的优选实施方式,所述步骤(2)中,超声振荡使固体产物、反应物b和溶剂b混合均匀。本发明还要求保护所述吲哚类化合物作为荧光探针在检测线粒体中的应用。本发明还要求保护所述吲哚类化合物作为荧光探针在检测线粒体中g-四链体dna中的应用。本发明所述吲哚类化合物作为荧光探针在体外实验中能够区分单链dna和双链dna、rna和线粒体中g-四链体dna,因此,所述吲哚类化合物具有良好的核酸区分性,可作为荧光探针检测线粒体,线粒体中g-四链体dna。本发明还要求保护所述吲哚类化合物在细胞成像中检测线粒体中的应用。本发明还要求保护所述吲哚类化合物在细胞成像中检测线粒体g-四链体dna中的应用。本发明所述吲哚类化合物作为荧光配体作用于细胞质内的线粒体,与线粒体中的核酸结合,可在在波长为405nm的光激发下成像,因此,所述吲哚类化合物荧光配体能够在细胞体系内对线粒体g-四链体dna进行成像和检测。本发明还要求保护所述吲哚类化合物在制备抗肿瘤药物中的应用。本发明所述吲哚类化合物对癌细胞具有比较大的细胞毒性,较低浓度即可抑制癌细胞的生长,因此,吲哚类化合物具有作为抗肿瘤药物的潜力。本发明还要求保护所述吲哚类化合物在线粒体功能调控方面的应用。本发明还要求保护所述吲哚类化合物在制备线粒体功能诊断试剂中的应用。本发明相对于现有技术,具有如下有益效果:本发明通过对吲哚结构的改造,引入苯乙烯基和氮杂环得到所述吲哚类化合物,所述吲哚类化合物是一种对线粒体g-四链体具有选择性结合的荧光配体,在体外实验中能够特异性识别线粒体g-四链体dna,在细胞实验中则能够对细胞中的线粒体进行染色,并且具有荧光强度高、结合常数强,检测限低等优点。另外,本发明所述吲哚类化合物配体能够调控并影响线粒体功能,具有发展成线粒体功能诊断试剂和抗肿瘤药物的潜力。附图说明图1为本发明实施例1制备的吲哚类化合物bym滴定不同类型核酸的荧光柱状图;图2为本发明实施例3制备的吲哚类化合物ym滴定不同类型核酸的荧光柱状图;图3为本发明实施例2制备的吲哚类化合物byp滴定不同类型核酸的荧光柱状图;图4为本发明实施例4制备的吲哚类化合物yp滴定不同类型核酸的荧光柱状图;图5为本发明实施例1制备的吲哚类化合物bym滴定线粒体g-四链体mt12086的荧光光谱中核酸浓度c与(f-f0)/f0拟合的曲线图;图6为本发明实施例3制备的吲哚类化合物ym滴定线粒体g-四链体mt12086的荧光光谱中核酸浓度c与(f-f0)/f0拟合的曲线图;图7为本发明实施例1制备的吲哚类化合物bym滴定不同浓度的线粒体g-四链体mt12086的荧光图;图8为本发明实施例3制备的吲哚类化合物ym滴定不同浓度的线粒体g-四链体mt12086的荧光图;图9为本发明实施例1制备的吲哚类化合物bym与线粒体g-四链体的结合能力拟合图;图10为本发明实施例3制备的吲哚类化合物ym与线粒体g-四链体的结合能力拟合图;图11为本发明实施例1制备的吲哚类化合物bym在hela细胞中成像图;图12为本发明实施例1制备的吲哚类化合物bym的浓度与对hela细胞抑制率的关系图;图13为本发明实施例2制备的吲哚类化合物byp的浓度与对hela细胞抑制率的关系图;图14为不同浓度的吲哚类化合物bym对cox-1的实时定量pcr结果图。具体实施方式为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。实施例1本实施例所述吲哚类化合物bym的制备,包括以下步骤:(1)称取0.4g(1.912mm)的1,1,2-三甲基-1h-苯并[e]吲哚置于防爆瓶中,4ml(42mm)环丁砜作为溶剂,超声使两者混合均匀,在通风橱条件下加入1,1,2-三甲基-1h-苯并[e]吲哚两倍摩尔量(3.824mm)的碘甲烷,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为24h;待反应体系冷却至室温后,加少量乙酸乙酯充分振荡,静置片刻,析出晶体,真空过滤,并用乙酸乙酯冲洗滤饼,烘干得到淡黄色晶体中间体a10.58g,薄板层析显示无副产物,中间体a1粗产率为86%;所述中间体a1的合成路线如下:(2)称取0.2g(0.569mm)的中间体a1和0.131mg(0.683mm)4-吗啡啉苯甲醛置于防爆瓶中,3ml(34.25mm)乙醇作为溶剂,超声片刻使混合均匀,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为12h;待反应体系冷却至室温后,会有固体析出,抽滤获得砖红色粉末,再通过柱层析对红色粉末中各组分进行分离,最终获得所述吲哚类化合物0.151g,计为bym,所述吲哚类化合物bym为暗红色晶体,产率为49%。目标产物吲哚类化合物bym的氢谱、碳谱数据如下:1hnmr(400mhz,dmso)δ8.40(t,j=12.6hz,2h),8.25(d,j=8.9hz,1h),8.18(d,j=8.2hz,1h),8.13(d,j=9.0hz,2h),8.02(d,j=8.9hz,1h),7.78(t,j=7.3hz,1h),7.67(t,j=7.5hz,1h),7.41(d,j=16.0hz,1h),7.13(d,j=9.0hz,2h),4.15(s,3h),3.80–3.72(m,4h),3.54–3.46(m,4h),2.00(s,6h).13cnmr(101mhz,dmso)δ181.74(s),154.85(s),153.09(s),140.06(s),137.21(s),133.91(s),133.18(s),131.12(s),130.48(s),128.72(s),127.29(s),127.00(s),124.53(s),123.35(s),113.97(s),113.38(s),107.20(s),66.28(s),53.40(s),46.89(s),34.52(s),26.23(s)。表明本发明实施例1成功吲哚类化合物bym。所述吲哚类化合物bym的合成路线如下:实施例2本实施例所述吲哚类化合物byp的制备,包括以下步骤:(1)所述中间体a1的制备方法与实施例1相同;(2)称取0.2g(0.569mm)的中间体a1和0.140mg(0.683mm)4-(4-甲基哌嗪)苯甲醛置于防爆瓶中,3ml(34.25mm)乙醇作为溶剂,超声片刻使混合均匀,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为12h;待反应体系冷却至室温后,会有固体析出,抽滤获得深桃红色粉末,再通过柱层析对红色粉末中各组分进行分离,最终获得吲哚类化合物0.240g,计为byp,产率为35%。所述吲哚类化合物byp为暗红色晶体。目标产物吲哚类化合物byp的氢谱、碳谱数据如下:1hnmr(400mhz,dmso)δ8.21–8.11(m,2h),8.01(d,j=9.0hz,1h),7.95(d,j=8.2hz,1h),7.89(d,j=9.0hz,2h),7.78(d,j=8.9hz,1h),7.54(t,j=7.2hz,1h),7.48–7.41(m,1h),7.17(d,j=16.0hz,1h),6.91(d,j=9.0hz,2h),3.93(d,j=12.8hz,3h),3.31(d,j=30.4hz,4h),2.37(d,j=55.1hz,4h),2.24–2.03(m,3h),1.78(d,j=10.4hz,6h).13cnmr(101mhz,dmso)δ181.92(s),153.89(s),152.93(s),140.04(s),137.37(s),133.81(s),133.24(s),131.14(s),130.48(s),128.74(s),127.17(d,j=18.7hz),124.91(s),123.38(s),114.56(s),113.44(s),107.73(s),53.50(s),45.08(s),34.69(s),26.18(s).表明本发明实施例2成功制备吲哚类化合物byp。所述吲哚类化合物byp的的合成路线如下:实施例3本实施例所述吲哚类化合物ym的制备,包括以下步骤:(1)称取0.4g(2.480mm)的2,3,3-三甲基-3h-吲哚置于防爆瓶中,4ml(42mm)环丁砜作为溶剂,超声片刻使两者混合均匀,将通风橱条件下加入两倍摩尔量(4.96mm)的碘甲烷,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为24h;待反应体系冷却至室温后,加乙酸乙酯充分振荡,静置片刻,析出晶体,真空过滤,并用乙酸乙酯冲洗滤饼,烘干得到淡粉色中间体a2晶体0.52g,薄板层析显示无副产物,中间体a2粗产率为70%;所述中间体a2的合成路线如下:(2)称取0.2g(0.664mm)的中间体a2和0.152mg(0.792mm)4-吗啡啉苯甲醛置于防爆瓶中,3ml(34.25mm)乙醇作为溶剂,超声片刻使两者混合均匀,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为12h;待反应体系冷却至室温后,会有固体析出,抽滤,得到红色粉末,再通过柱层析对红色粉末中各组分进行分离,最终获得吲哚类化合物0.148g,计为ym,产率为45%;所述吲哚类化合物ym为暗红色粉末。目标产物吲哚类化合物ym的氢谱、碳谱数据如下:氢谱数据:1hnmr(400mhz,dmso)δ8.33(d,j=15.9hz,1h),8.11(d,j=9.0hz,2h),7.79(dd,j=18.0,7.4hz,2h),7.56(ddd,j=21.8,10.9,6.9hz,2h),7.37(d,j=15.9hz,1h),7.12(d,j=9.1hz,2h),4.02(s,3h),3.82–3.69(m,4h),3.59–3.46(m,4h),1.76(s,6h).。碳谱数据:13cnmr(101mhz,dmso)δ180.82(s),154.96(s),154.18(s),143.32(s),142.48(s),134.11(s),129.23(s),128.52(s),124.45(s),123.16(s),114.50(s),113.91(s),107.45(s),66.27(s),51.69(s),46.86(s),33.96(s),26.50(s)。表明本发明实施例3成功制备吲哚类化合物ym。吲哚类化合物ym的合成路线如下:实施例4本实施例所述吲哚类化合物yp的制备,包括以下步骤:(1)所述中间体a1的制备方法与实施例3相同;(2)称取0.2g(0.664mm)的中间体a2和0.162mg(0.792mm)4-(4-甲基哌嗪)苯甲醛置于防爆瓶中,3ml(34.25mm)乙醇作为溶剂,超声片刻使两者混合均匀,将反应体系置于油浴锅中并打开磁力搅拌,反应温度为60℃,反应时间为12h;待反应体系冷却至室温后,会有固体析出,抽滤,获得红色粉末,再通过柱层析对红色粉末中各组分进行分离,最终获得吲哚类化合物0.113g,计为yp,产率为36%,所述吲哚类化合物yp为暗红色晶体;目标产物吲哚类化合物yp的氢谱、碳谱数据如下:氢谱数据:1hnmr(400mhz,dmso)δ8.31(d,j=15.8hz,1h),8.09(d,j=9.0hz,2h),7.77(dd,j=20.0,7.3hz,2h),7.55(tdd,j=14.8,10.8,4.2hz,2h),7.37–7.31(m,1h),7.11(d,j=9.1hz,2h),4.01(s,3h),3.58–3.51(m,4h),2.47–2.42(m,4h),2.23(d,j=7.1hz,3h),1.76(s,6h).。碳谱数据:13cnmr(101mhz,dmso)δ180.63(s),154.81(s),154.20(s),143.25(s),134.30(s),129.21(s),128.38(s),124.02(s),123.14(s),114.38(s),114.01(s),107.01(s),54.72(s),51.60(s),46.60(s),46.07(s),33.87(s),26.55(s).。吲哚类化合物yp的合成路线如下:效果例1本效果例为所述吲哚类化合物对不同核酸选择性的荧光光谱实验。测试样品:实施例1-4所制备的吲哚类化合物;测试方法:将5mm的测试样品分别使用含有60mmk+的tris-hcl缓冲液稀释成1μm的浓度,再加入不同种类的核酸,使用荧光分光光度计测试荧光强度;各测试样品的荧光分光光度计参数为:各样品的狭缝宽度均为10,扫描速度均为800nm;吲哚类化合物bym:ex=505nm;吲哚类化合物ym:ex=497nm,吲哚类化合物byp:ex=494nm;吲哚类化合物yp:ex=484nm;所述核酸包括:rna;dt21的2种单链dna;ds26、ds12、4at3种双链dna;mt12086、mt16250、mt1015、mt6363、mt377、mt8095、mt10252、mt9438、mt7148种线粒体g-四链体dna,具体序列如下表1。表1本发明所述核酸的种类、序列及类别信息图1、2、3和4分别为本发明所述吲哚类化合物bym、吲哚类化合物ym、吲哚类化合物byp和吲哚类化合物yp滴定不同类型核酸的荧光柱状图;从图结果可以看出,四种吲哚类化合物在体外实验中均能够区分单链dna和双链dna、rna和线粒体g-四链体dna,因此,所述吲哚类化合物具有良好的核酸区分性。效果例2本效果例为所述吲哚类化合物的灵敏度测试。测试样品:实施例1和实施例3所制备的样品;测试方法:将5mm的测试样品稀释成1μm的浓度,再使用荧光分光光度计(光分光光度计的测试参数:狭缝宽度=10,扫描速度=200nm,化合物bym:ex=515nm;化合物ym:ex=497nm)扫描,然后慢慢加入线粒体g-四链体dna(mt12086)做到使其饱和;检测限的计算公式:lod=k×sb/mlod为化合物结合常数,m是dna的浓度c与(f-f0)/f0的所做直线的斜率,sb为用仪器空白多次测量的空白偏差,k值按照国际纯粹和应用化学联合会所建议通常取为3。图5和6分别为吲哚类化合物bym和吲哚类化合物yb荧光配体滴定mt12086的荧光光谱中的mt12086的浓度与f-f0)/f0拟合的曲线,从图中可以看出,吲哚类化合物bym滴定mt12086的荧光光谱中mt12086的浓度与f-f0)/f0的所做直线的斜率为0.993;吲哚类化合物ym滴定mt12086的荧光光谱中mt12086的浓度与f-f0)/f0的所做直线的斜率为0.993。因此,根据检测限的计算公式计算吲哚类化合物bym的检测限lod为6.30nmol/l;吲哚类化合物ym的检测限lod为11.84nmol/l,都具有较低检测限,说明所述吲哚类化合物荧光配体有较高的灵敏度,具有良好的商业价值。图7和8分别为吲哚类化合物bym和吲哚类化合物yb滴定不同浓度的线粒体g-四链体mt12086荧光图;图7和图8中各荧光曲线代表不同浓度的线粒体g-四链体mt12086的荧光曲线;图7中的荧光曲线自下而上分别为吲哚类化合物bym滴定0、0.071、0.14、0.21、0.29、0.36、0.43、0.57、0.71、0.86、1、1.14、1.28、1.43、1.57、1.71、1.86μmol/l浓度线粒体g-四链体mt12086的荧光曲线;图8中的荧光曲线自下而上分别为吲哚类化合物yb滴定0、0.071、0.14、0.21、0.29、0.36、0.43、0.57、0.71、0.86、1、1.14、1.28、1.43、1.57、1.71、1.86μmol/l浓度线粒体g-四链体mt12086的荧光曲线。从图7和8中可以看出,随着dna浓度的增大,荧光强度也增大。效果例3本效果例为所述吲哚类化合物与线粒体g-四链体的结合能力研究。测试样品:实施例1和实施例3所制备的样品;测试方法:小分子荧光配体与g-四链体dna的结合能力kd值是衡量荧光配体作为g-四链体dna荧光配体的重要指标。通过图9和10的吲哚类化合物与线粒体g-四链体的结合能力拟合图和表2和3的数据,其中图9和图10中横坐标[dna]/[ligand]代表核酸与测试样品浓度比:cdna/cligand。计算所述吲哚类化合物与g-四链体dna相互作用的结合能力,计算公式如下:f/f0=1+(q-1)/2{n+1+x-[(x+1+n)24x]1/2}其中,f0代表不加入核酸时化合物的荧光强度,f代表加入核酸时化合物的荧光强度,fmax代表滴定饱和时的荧光强度,n=(kacligand)-1,q=fmax(f0)-1,x=ncdna(cligand)-1,ka=kd-1;cligand代表测试样品的浓度;cdna代表核酸的浓度。表2吲哚类化合物bym与线粒体g-四链体的结合能力参数表3吲哚类化合物ym与线粒体g-四链体的结合能力参数结合表2、表3和图9、10的结果,经计算,吲哚类化合物bym与mt12086g-四链体为13.16*105m-1;吲哚类化合物yb与mt12086g-四链体2.22*105m-1,具有较高结合能。效果例4本效果例为所述吲哚类化合物的细胞成像实验。测试样品:实施例1和实施例3所制备的样品;测试方法:(1)细胞染色:将宫颈瘤细胞(hela)细胞接种在2个细胞培养皿中,使细胞的密度约为5000个/ml,然后在37℃、5%co2环境下培养70h。接着弃去培养皿中的细胞培养液,用预冷的1×pbs洗3次,然后再加入预冷的纯甲醇1.5ml常温避光放置5min。弃去上步培养皿中的溶液,用预冷的1×pbs洗3次,在上述培养皿中分别加入1ml、5um的吲哚类化合物bym,1ml、8um的吲哚类化合物ym,然后放置30min。弃去上步6孔板中的化合物溶液,用预冷的1×pbs洗3次,在上述培养皿中加入5um的hoehst溶液1ml并37℃放置15min,然后再用预冷的1×pbs洗6次,每次浸泡5min,最后在激光共聚焦显微镜下观察细胞染色情况。(2)dna酶与rna酶消化:将宫颈瘤细胞(hela)细胞接种在3个细胞培养皿中,使细胞的密度约为5000个/ml,然后在37℃、5%co2环境下培养24h。接着弃去培养皿中的细胞培养液,用预冷的1×pbs洗3次,然后再加入预冷的纯甲醇1.5ml常温避光放置5min。弃去上步培养皿中的溶液,用预冷的1×pbs洗3次,在上述培养皿中分别加入1ml、5μm的吲哚类化合物bym,然后放置30min。弃去上步6孔板中的化合物溶液,用预冷的1×pbs洗3次,在上述培养皿中加入5μm的hoehst溶液1ml并37℃放置15min,然后再用预冷的1×pbs洗6次,每次浸泡5min。在一个培养皿中加入dna酶进行消化,一个皿中加入rna酶进行消化,最后在激光共聚焦显微镜下观察细胞染色情况。图11为本发明实施例1制备的吲哚类化合物bym在hela细胞中成像图,图11(a)为在波长为405nm的光激发下实施例1制备的吲哚类化合物bym在hela细胞中成像图;图11(b)为在波长为405nm的光激发下实施例1制备的吲哚类化合物bym经dna酶消化后在hela细胞中成像图;图11(c)为在波长为405nm的光激发下实施例1制备的吲哚类化合物bym经rna酶消化后在hela细胞中成像图;图11(d)为在波长为505nm的光激发下实施例1制备的吲哚类化合物bym在hela细胞中成像图;图11(d)为在波长为505nm的光激发下实施例1制备的吲哚类化合物bym经dna酶消化后在hela细胞中成像图;图11(f)为在波长为505nm的光激发下实施例1制备的吲哚类化合物bym经rna酶消化后在hela细胞中成像图;图11(g)为明视场下实施例1制备的吲哚类化合物bym在hela细胞中成像图;图11(h)为明视场下实施例1制备的吲哚类化合物bym经dna酶消化后在hela细胞中成像图;图11(i)为明视场下实施例1制备的吲哚类化合物bym经rna酶消化后在hela细胞中成像图;图11(j)为图11(a)和图11(d)合并得到的吲哚类化合物bym在hela细胞中成像图;图11(k)为图11(b)和图11(e)合并得到的吲哚类化合物bym经dna酶消化后在hela细胞中成像图;图11(l)为图11(c)和图11(f)合并得到的吲哚类化合物bym经rna酶消化后在hela细胞中成像图;从图11中可以看出,吲哚类化合物bym配体与细胞核染料hoehst复染hela细胞的细胞成像图,吲哚类化合物bym荧光配体作用于细胞质内,真核细胞中细胞质中只有线粒体携带dna,其他细胞器没有,并且经过dna酶消化后,荧光消失,而经过rna酶消化的细胞荧光没有明显变化,由此可以证明,化合物bym染色位置为线粒体。化合物bym与细胞质(线粒体)中的核酸结合,该系荧光配体能够在细胞体系内对线粒体g-四链体dna进行成像和检测。效果例5本效果例为所述吲哚类化合物对hela细胞毒性实验。测试样品:实施例1和实施例2所制备的样品;测试方法:(1)将宫颈瘤细胞(hela)接种至96孔板中培养,使每孔细胞数量为3000个,然后在37℃、5%co2环境下培养;24h后细胞贴壁,弃去96孔板中的培养基,加入用培养基配置的浓度为20、10、5、2.5、1.25μm的吲哚类化合物bym溶液,孵育48h后,弃去含有化合物的培养基,每孔100μl的0.5mg/ml的mtt,孵育4h后,弃去mtt溶液,每孔加入100μldmso,置于摇床上孵育20min,用酶标仪测量其吸光度值,测量波长为570nm。(2)将宫颈瘤细胞(hela)接种至96孔板中培养,使每孔细胞数量为3000个,然后在37℃、5%co2环境下培养;24h后细胞贴壁,弃去96孔板中的培养基,加入用培养基配置的浓度为20、10、5、2.5、1.25μm的吲哚类化合物byp溶液,孵育48h后,弃去含有化合物的培养基,每孔100μl的0.5mg/ml的mtt,孵育4h后,弃去mtt溶液,每孔加入100μldmso,置于摇床上孵育20min,用酶标仪测量其吸光度值,测量波长为570nm。细胞存活率与吲哚类化合物浓度的关系见图12和13,细胞的半数抑制浓度(ic50)见表4。表4吲哚类化合物bym、吲哚类化合物byp对hela细胞的ic50从图12、13与表4可以看出,吲哚类化合物bym与吲哚类化合物byp对宫颈癌细胞(hela)具有比较大的细胞毒性,较低浓度即可抑制癌细胞的生长,由此可以看出吲哚类化合物bym与byp具有作为抗肿瘤药物的潜力。效果例6本效果例为所述吲哚类化合物对线粒体g-四链体的调控作用。测试样品:实施例1所制备的样品;测试方法:(1)将宫颈瘤细胞(hela)接种至6孔板中培养,使每孔细胞数量为10000个培养24h后,弃去细胞原有的培养液,加入用培养基配置的0、1、2、5、10、20μm的吲哚类化合物bym,培养48h后,弃去细胞原有的培养液,并用pbs进行清洗。接着每孔加入1mltrizol试剂,在冰袋上完成用trizol试剂吹打整个皿的底部,吹打完成后将其转移如1.5mlep管中,然后室温下静置10min。然后往每个ep管中加入200μl三氯甲烷,手摇15s,室温静置3min,紧接着4℃,12000r离心15分钟,离心后,转移上层清液转移到ep管中,然后加入500μl异丙醇,混匀,室温静置10min,紧接着4℃,12000r离心10分钟后,弃去上清,保留ep管中的白色沉淀。加入1ml75%乙醇洗涤rna沉淀,上下摇晃后,4℃,7500r离心5分钟,弃去上清,将ep管倒扣在吸水纸上,再敞口放置使乙醇挥发,最后加入30~50μldepc水,溶解rna,涡旋。测量rna浓度,并使用depc水将6个样品调整至浓度一致。(2)将步骤(1)得到的6个样品,分别加入25μl1×pcr缓冲液,2μm引物(序列见表5),0.16mmdntp和2.5utaq聚合酶的溶液中来进行pcr终止试验。将反应混合物在热循环仪中在以下循环条件下孵育:94℃5min,94℃30s,30个循环,56℃30s和72℃30s的30个循环。将扩增的产物在20%聚丙烯酰胺凝胶上分离,并用sybrgold染色。表5引物序列引物序列(5’-3’)mtcox1-fgcctgactggcattgtattamtcox1-rggttcgattccttccttttt从图14为不同浓度的吲哚类化合物bym对cox-1的实时定量pcr结果图,所述pcr为聚合酶链式反应,图中0、5、10、20分别为吲哚类化合物bym浓度为0μm、5μm、10μm、20μm。从图中可以看出随着吲哚类化合物bym浓度增加,线粒体当中的cox-1基因的表达量逐渐下降,这说明吲哚类化合物bym可能通过作用于线粒体g-四链体,并影响了cox-1基因的表达,因此,本发明所述吲哚类化合物具有调控线粒体功能的潜力。最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1