一种以烯烃和甲醇为原料合成1,3-二元醇的方法与流程
本发明属于化工产品的化学合成领域,具体而言涉及一种耦合脱氢/缩合/水解过程制备1,3-二元醇的方法。
背景技术:
1,3-二元醇在医药、化工、材料、燃料等领域有重要的用途。如在材料工业中,1,3-丁二醇可作为聚酯、聚氨酯合成单体以及溶剂、抗冻剂等。
制备1,3-丁二醇的方法已经有很多文献报道,比较常用的是以乙醛为主要原料进行缩合的方法制备1,3-丁二醇。如专利us5345004就公开了三步法制备1,3-丁二醇,首先是乙醛与醇缩合制备2,6-二甲基-1,3-二噁烷-4-醇,第二步将上述产物分解为二聚间羟丁醛,然后经过氢化得到1,3-丁二醇。另外就是以烯烃与醛的缩合反应制备1,3-二元醇的方法。甲醛首先在酸催化下形成甲醛碳正离子,甲醛碳正离子再进攻烯烃的c=c双键形成烯醇碳正离子,烯醇碳正离子会继续与甲醛发生成环反应,生成1,3-二噁烷,然后在酸性水溶液中发生水解生成1,3-二元醇。cn201210342364.9,cn201410353020.7,cn201410353633.0,cn201710225481.x,cn201810411956.9,cn200810150864.6,cn201410774243.0,cn201711111544.5各自公开了以液体酸、固体酸(包括分子筛、氧化铝、酸性金属复合物、离子交换树脂、杂多酸等)、离子液体等为催化剂促进丙烯与甲醛缩合制备甲基-1,3-二噁烷,以及甲基-1,3-二噁烷水解制备1,3-丁二醇的方法。上述方法或者以甲醛和烯烃为原料经过普林斯缩合反应制备甲基-1,3-二噁烷,或者将普林斯缩合反应与后续的水解反应耦合一步得到1,3-丁二醇(因为普林斯缩合和水解反应均为酸催化)。目前上述反应所使用的原料甲醛主要为甲醛水溶液(38%)、多聚甲醛或三聚甲醛。甲醛的工业化生产方法为甲醇脱氢/氧化,反应温度普遍较高(400℃以上)。利用金属活性组分高度分散的负载型金属催化剂具有的超高反应活性,甲醇可以在较低的反应温度下发生脱氢反应生成甲醛(acscatal.2019,9,10977-10982,cn200510133886.8)。
然而上述现有技术往往都是从甲醛作为起始反应物经过多步反应才能最终制备1,3-二元醇,反应步骤复杂,成本较高。特别是甲醛的工业化生产方法为甲醇脱氢/氧化,反应温度通常为400℃至600℃。而普林斯缩合反应的反应温度通常不超过250℃,一般在120℃至200℃之间,这就造成甲醇脱氢反应和普林斯缩合反应很难一次性完成,因此,现有技术往往只能通过多步法进行。
技术实现要素:
针对上述现有技术存在的问题,本发明以甲醇和烯烃为原料,采用金属活性组分高度分散的负载型多功能催化剂,将脱氢/普林斯缩合/水解三个步骤耦合一步制备1,3-二元醇。
根据本发明的一个方面,本发明的一个目的在于提供一种以烯烃和甲醇为原料,经过一步反应制备1,3-二元醇的方法。该方法原料成本低,步骤简单,可连续生产。
根据本发明的所述制备1,3-二元醇的方法,所述方法包括如下步骤:
1)在反应器中加入催化剂,在氢氮混合气氛下升温至反应温度100-350℃还原30min-6h。还原结束后切换至氮气气氛后降温至60-180℃,保持反应器内压力0.5-8mpa反应。
2)将烯烃和甲醇水溶液分别通入反应器中进行反应,以甲醇计空速为0.01-10h-1。
3)反应产物经过冷凝和气液分离后进入产品储罐。
4)将步骤3)中得到的反应产物进行精馏分离分离,得到纯度大于99%的1,3-二元醇产品。
优选地,所述步骤1)中所述反应器选自固定床反应器。
优选地,所述步骤1)中所述氢氮混合气中氢气的体积为1-25%,氮气的体积为75-99%。
优选地,所述步骤1)中所述催化剂为金属活性组分高度分散的负载型金属催化剂。优选地,所述步骤1)中所述催化剂的金属活性组分包括pd、pt、ru、rh、ir、ag、ni、co和cu中的至少一种;所述催化剂的载体为分子筛,包括h-zsm-5、h-zsm-11、h-x、h-y、h-beta、h-mor、sapo-11、sapo-34中的至少一种。
优选地,所述步骤1)中所述催化剂的金属活性组分包括pd、pt、ru、rh、ir和cu中的至少一种,更优选为ru、cu和ir中的至少一种。
优选地,所述步骤1)中所述催化剂中金属活性组分的负载量为0.01wt-1.5wt%。更优选为0.05wt-0.7wt%。
优选地,所述步骤2)中所述甲醇与烯烃的摩尔比为1:0.1-1:10,进一步优选为1:0.5-1:5,最优选为1:0.5-1:2。
优选地,所述步骤2)中所述烯烃包括乙烯、丙烯、异丁烯、苯乙烯和环己烯。
优选地,所述步骤2)中所述甲醇水溶液中水的质量浓度为0-80wt%,优选为30-90wt%,水的质量浓度为0时表示使用纯甲醇。
优选地,所述步骤2)中甲醇计空速为0.05-5h-1,更优选为0.05-1h-1,更优选为0.05-0.2h-1。
优选地,所述步骤1)中所述催化剂按照如下方法制备:
a1)将二乙烯三胺和金属活性组分的盐先后加入水中,搅拌溶解。
b1)将四丙基氢氧化铵和重量百分百浓度为96%的硫酸溶解在水中搅拌10min,将铝酸钠和氢氧化钠溶解在水中搅拌10min,将上述两种溶液加入硅溶胶中,混合均匀然后向其中加入步骤a1)中得到的溶液,持续剧烈搅拌后,放入不锈钢反应釜进行高温水热晶化。
c1)晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后干燥,再在500℃焙烧。
d1)将步骤c1)中焙烧后的催化剂按照固体和液体质量比为1:20加入到1.0mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后干燥,再在500℃焙烧。
e1)然后用高纯氢气将步骤d1)中焙烧后的催化剂在500℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂。
其中,优选地,所述金属活性组分的盐选自活性金属的盐酸盐、硝酸盐、硫酸盐等,优选为盐酸盐和硝酸盐。
优选地,所述金属活性组分包括pd、pt、ru、rh、ir、ag、ni、co和cu中的至少一种;优选地,所述金属活性组分包括pd、pt、ru、rh、ir和cu中的至少一种,更优选为ru、cu和ir中的至少一种。
所述金属盐和二乙烯三胺的质量比为1:3-1:20,更优选为1:3-1:10。
所述四丙基氢氧化铵与硫酸的质量比为1:1-1-3,所述铝酸钠与氢氧化钠的质量比为1:2-1:7;所述四丙基氢氧化铵在水中的质量浓度为5wt%-15wt%;所述铝酸钠在水中的质量浓度为0.5wt%-5wt%;所述四丙基氢氧化铵与铝酸钠的质量比为10:1-5:1;所述硅溶胶与铝酸钠的质量比为85:1-40:1.
所述水热晶化的温度为120-200℃,更优选为140-180℃。
所述水热晶化的时间为15-60h,更优选为20-30h。
优选地,所述催化剂中金属活性组分的负载量为0.01wt-1.5wt%。更优选为0.01wt-0.7wt%。
或者,所述步骤1)中所述催化剂按照如下方法制备:
a2)将乙醇、乙二胺和水杨醛混合,在室温下搅拌反应1h,逐渐析出黄绿色沉淀,过滤后60℃干燥5h得到黄绿色salen结晶。
b2)将铝酸钠和氢氧化钠加入水中,剧烈搅拌溶解后,向其中加入步骤a2)制备的salen结晶和金属活性组分的盐,继续搅拌后将溶液加入硅溶胶中,在室温下陈化2天后再进行水热晶化。
c2)晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后100℃下干燥24h,再在500℃下焙烧6h,也可在高真空(真空度1torr)下150℃焙烧6h。
d2)将步骤c2)中制备的产物按照固体和液体质量比为1:20加入到1.0mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后在100℃下干燥24h,再在500℃焙烧6h,也可在高真空(真空度1torr)下150℃焙烧6h。
e2)然后用高纯氢气将步骤d2)中焙烧后的催化剂在200℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂。
其中,优选地,所述金属活性组分的盐选自盐酸盐、硝酸盐、硫酸盐等,优选为盐酸盐和硝酸盐。
优选地,所述金属活性组分包括pd、pt、ru、rh、ir、ag、ni、co和cu中的至少一种;优选地,所述金属活性组分包括pd、pt、ru、rh、ir和cu中的至少一种,更优选为ru、cu和ir中的至少一种。
优选地,所述催化剂中金属活性组分的负载量为0.01wt-1.5wt%。更优选为0.01wt-0.7wt%。
所述乙二胺与水杨醛的质量比为1:2-1:10,更优选为1:2-1:5。
所述乙二胺与乙醇的质量比为1:20-1:60,更优选为1:30-1:45。
所述铝酸钠与氢氧化钠的质量比为1:0.5-1:5,更优选为1:1-1:3。所述铝酸钠与硅溶胶的质量比为1:5-1:15,更优选为1:6-1:8。
所述铝酸钠与水的质量比为1:3-1:10,更优选为1:4-1:7。
所述金属盐与salen的质量比为1:2-1:10,更优选为1:2-1:5。
所述水热晶化的温度为80-200℃,更优选为80-160℃。
有益效果
本发明提供的1,3-二元醇的合成方法原料成本低,可连续生产,过程效率高简单易行,经济无污染,易于实现工业化生产。
说明书附图
为了更清楚地说明本发明具体实施方式或现有技术中的技术方案,下面将对具体实施方式或现有技术描述中所需要使用的附图作简单的介绍,显而易见地,下面描述中的附图是本发明的一些实施方式,对本领域普通技术人员而言,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
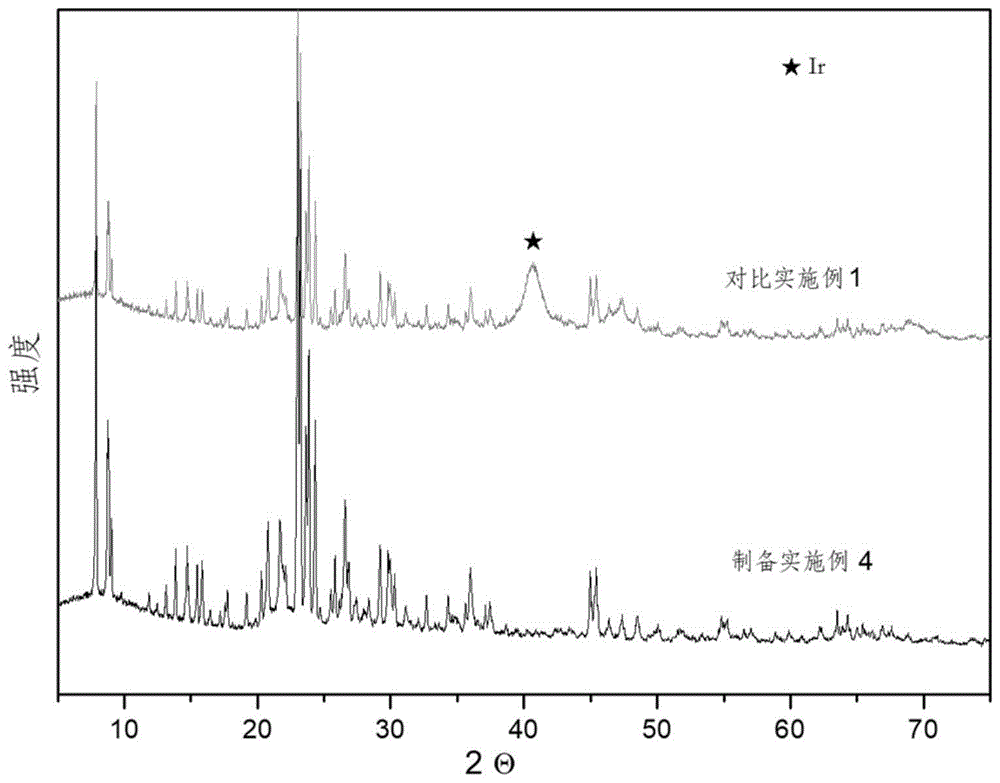
图1为制备实施例4与对比制备实施例1中制备的催化剂的xrd谱图;
图2为制备实施例4与对比制备实施例1中制备的催化剂的tem照片(左侧:制备实施例4,右侧:对比制备实施例1)。
图3为实施例4中反应产物的气相色谱图。
图4为实施例4中主要反应产物的质谱检测谱图。
具体实施方式
以下,将详细地描述本发明。在进行描述之前,应当理解的是,在本说明书和所附的权利要求书中使用的术语不应解释为限制于一般含义和字典含义,而应当在允许发明人适当定义术语以进行最佳解释的原则的基础上,根据与本发明的技术方面相应的含义和概念进行解释。因此,这里提出的描述仅仅是出于举例说明目的的优选实例,并非意图限制本发明的范围,从而应当理解的是,在不偏离本发明的精神和范围的情况下,可以由其获得其他等价方式或改进方式。
以下实施例仅是作为本发明的实施方案的例子列举,并不对本发明构成任何限制,本领域技术人员可以理解在不偏离本发明的实质和构思的范围内的修改均落入本发明的保护范围。除非特别说明,以下实施例中使用的试剂和仪器均为市售可得产品。
本发明中所使用甲醇、四丙基氢氧化铵、铝酸钠、乙二胺、二乙烯三胺、水杨醛、氯化铜采购自国药集团化学试剂有限公司;氯化铱、氯化钌采购自陕西瑞科新材料有限公司;丙烯采购自青岛德海伟业科技有限公司;对比实施例中所用h-zsm-5采购自天津南化催化剂有限公司;去离子水为自制;固定床反应器为自制;气相色谱仪为岛津公司的shimazu-gc2010plus,气质联用仪为岛津公司的shimazu-qp-2010ultra。
根据本发明的制备1,3-二元醇的方法包括如下步骤:
1)在反应器中加入催化剂,在氢气气氛下升温至反应温度100-350℃还原30min-6h。还原结束后切换至氮气气氛后降温至60-180℃,反应器内保持0.5-8mpa反应。
2)将烯烃和甲醇水溶液分别通入反应器中进行反应,以甲醇计反应空速为0.01-10h-1。
3)反应产物经过冷凝和气液分离后进入产品收集罐。
在根据本发明的制备1,3-二元醇的方法中,以烯烃和甲醇为原料,经过脱氢/缩合/水解反应得到1,3-二元醇。产物过0.22μm滤膜,用气相色谱(gc)进行分析检测。气相色谱检测条件:仪器:岛津gc2010plus,色谱柱:intercap-ffap,30m×0.25mm×0.25um,汽化室温度250℃,fid温度300℃,柱温箱升温程序:60℃保持1min,然后以15℃/min速度升温至230℃保持10min。通过气质联用(gc-ms)和标准物gc保留时间对照对产物进行定性分析。用气相色谱对产物进行定量测定,通过与标准物保留时间和峰面积大小比对进行定量分析。液体产物的收率以(目标产物的摩尔量)/(甲醇的摩尔量)×100%进行计算,相关计算公式如下:
甲醇的转化率(%)=(n甲醇1-n甲醇2)/n甲醇1×100%
其中n甲醇1为反应前甲醇的摩尔量,n甲醇2为反应后甲醇的摩尔量;
1,3-二元醇的收率(%)=(n二元醇/n甲醇1)×100%
其中n甲醇1为反应前甲醇的摩尔量,n二元醇为生成的二元醇的摩尔量;
1,3-二元醇的选择性(%)=二元醇的收率/甲醇的转化率×100%
其他副产物收率与选择性参考1,3-二元醇的计算方法。
制备实施例1
在10g水中先后加入1g二乙烯三胺和0.24g氯化钌,搅拌溶解得到溶液a。
将8.0g四丙基氢氧化铵和6.2g质量百分百浓度为96%的硫酸溶解在100g水中搅拌10min,将0.9g铝酸钠和5.9g氢氧化钠溶解在水中搅拌10min,将上述两种溶液加入60g硅溶胶(质量含30%的sio2)中,混合均匀然后向其中加入溶液a,持续剧烈搅拌30min后,放入不锈钢反应釜进行150℃水热晶化24h。
晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后在100℃下干燥24h,再在马弗炉中以2℃/min的升温速率升温至500℃焙烧6h。将上述催化剂按照固体和液体质量比为1:20加入到1.0mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后在100℃下干燥24h,再在马弗炉中以2℃/min的升温速率升温至500℃焙烧6h。然后用高纯氢气在200℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂1。
制备实施例2
在10g水中先后加入1g二乙烯三胺和0.24g氯化钌,搅拌溶解得到溶液a。
将8.0g四丙基氢氧化铵和6.2g质量百分百浓度为96%的硫酸溶解在100g水中搅拌10min,将0.9g铝酸钠和5.9g氢氧化钠溶解在水中搅拌10min,将上述两种溶液加入60g硅溶胶(质量含30%的sio2)中,混合均匀然后向其中加入溶液a,持续剧烈搅拌30min后,放入不锈钢反应釜进行150℃水热晶化24h。
晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后在100℃下干燥24h,高真空下(1torr)以2℃/min的升温速率升温至150℃焙烧6h。将上述催化剂按照固体和液体质量比为1:20加入到1mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后在100℃下干燥24h,再在高真空下(1torr)以2℃/min的升温速率升温至150℃焙烧6h。然后用高纯氢气在200℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂2。
制备实施例3
在烧瓶中加入30g乙醇,0.7g乙二胺,2g水杨醛,在室温下搅拌反应1h,溶液中会逐渐有黄绿色沉淀析出,过滤后60℃干燥5h得到黄绿色salen结晶。在14g水中加入2.7g铝酸钠,2g氢氧化钠,剧烈搅拌溶解后,向其中加入0.1gsalen和43mg氯化铱,继续搅拌10min后,将上述溶液加入20g硅溶胶中,在室温下陈化2天后再转入带有聚四氟内衬的水热釜中在95℃下晶化3天。
晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后在100℃下干燥24h,再在马弗炉中以2℃/min的升温速率升温至500℃焙烧6h。将上述催化剂按照固体和液体质量比为1:20加入到1mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后在100℃下干燥24h,再在马弗炉中以2℃/min的升温速率升温至500℃焙烧6h。然后用高纯氢气在200℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂3。
制备实施例4
在烧瓶中加入30g乙醇,0.7g乙二胺,2g水杨醛,在室温下搅拌反应1h,溶液中会逐渐有黄绿色沉淀析出,过滤后60℃干燥5h得到黄绿色salen结晶。在14g水中加入2.7g铝酸钠,2g氢氧化钠,剧烈搅拌溶解后,向其中加入0.1gsalen和43mg氯化铱,继续搅拌10min后,将上述溶液加入20g硅溶胶中,在室温下陈化2天后再转入带有聚四氟内衬的水热釜中在95℃下晶化3天。
晶化结束后离心,将得到的固体用去离子水洗涤至滤液呈中性后在100℃下干燥24h,再在高真空下(1torr)以2℃/min的升温速率升温至150℃焙烧6h。将上述催化剂按照固体和液体质量比为1:20加入到1mol/l的硝酸铵溶液中,在90℃水浴中恒温搅拌8h进行离子交换,交换结束后在100℃下干燥24h,再在高真空下(1torr)以2℃/min的升温速率升温至150℃焙烧6h。然后用高纯氢气在200℃下还原6h,在高纯氮气吹扫下降至室温继续吹扫12h后得到催化剂4。
制备实施例5
除了采用0.43g氯化铱代替0.24g氯化钌以外,按照实施例1相同的方式制备催化剂5。
制备实施例6
除了采用0.20g氯化铜代替0.24g氯化钌以外,按照实施例1相同的方式制备催化剂6。
对比制备实施例1
在烧杯中加入25mg氯化铱,加入100g去离子水搅拌溶解。向上述溶液中加入20gh-zsm-5分子筛(采购自天津南化催化剂有限公司),继续在室温下搅拌混合24h。将上述混合物旋蒸除去大部分水后在120℃干燥12h。将干燥后的粉末置于管式炉中,通入高纯氢气在500℃下还原2h,然后在高纯氮气吹扫下降至室温继续吹扫12h后得到对比催化剂。
图1为制备实施例4与对比制备实施例1中制备的催化剂的xrd谱图;
图2为制备实施例4与对比制备实施例1中制备的催化剂的tem照片(左侧:制备实施例4,右侧:对比制备实施例1)。
可以看出,制备实施例很好的合成了zsm5分子筛,但是对比实施例的xrd谱图上有明显的金属ir衍射峰,而制备实施例的xrd谱图上看不到金属ir的衍射峰,这说明制备实施例中所述催化剂上金属ir具有较高的分散度。从tem对比结果可以看出,相比于制备实施例所述催化剂,对比实施例的催化剂tem结果可以看出明显的ir金属颗粒,金属颗粒粒径较大且分布不均匀,而制备实施例上的金属颗粒很小,这与xrd检测的结果是相一致的。这说明相比于对比实施例,制备实施例中所述催化剂上的金属活性组分的分散程度高,因此其具有较高的甲醇低温反应活性,可以实现低温下甲醇的脱氢生成甲醛,生成的甲醛在酸催化剂的作用下与烯烃发生普林斯缩合反应与水解反应得到二元醇。
实施例1
采用制备实施例1制备的催化剂进行试验。
1)在固定床反应器中加入10g催化剂1,在氢氮混合气(体积:5%h2-95%n2)下以3℃/min的速度升温至200℃还原3h后切换为氮气气氛,然后降温至100℃,用氮气将体系压力调整为6mpa。
2)用柱塞泵分别将丙烯(液体)和甲醇水溶液(90%)通入反应器中进行反应,以甲醇计反应空速为0.1h-1,甲醇与丙烯的摩尔比为1:1。
3)反应产物经过冷凝和气液分离后收集,收集到的产物进行gc(气相色谱)检测后得甲醇的转化率为93.3%,主要产物包括1,3-丁二醇(选择性79.2%),4-甲基-1,3-二噁烷(选择性3.2%),四氢吡喃-4-醇(选择性3.6%)和二甲醚(选择性9.5%)。
实施例2
采用制备实施例2制备的催化剂进行试验。
1)在固定床反应器中加入10g催化剂2,在氢氮混合气(体积:5%h2-95%n2)下以3℃/min的升温速度升温至200℃还原3h后切换为氮气气氛,然后降温至100℃,压力6mpa。
2)用柱塞泵分别将丙烯(液体)和甲醇水溶液(90%)通入反应器中进行反应,以甲醇计反应空速为0.1h-1,甲醇与丙烯的摩尔比为1:1。
3)反应产物经过冷凝和气液分离后收集,收集到的产物进行gc检测后得甲醇的转化率为90.0%,主要产物包括1,3-丁二醇(选择性72.3%),4-甲基-1,3-二噁烷(选择性4.1%),四氢吡喃-4-醇(选择性4.6%)和二甲醚(选择性13.5%)。
实施例3
采用制备实施例3制备的催化剂进行试验。
1)在固定床反应器中加入10g催化剂3,在氢氮混合气(体积:5%h2-95%n2)下以3℃/min的升温速度升温至200℃还原3h后切换为氮气气氛,然后降温至100℃,压力6mpa。
2)用柱塞泵分别将丙烯(液体)和甲醇水溶液(90%)通入反应器中进行反应,以甲醇计反应空速为0.1h-1,甲醇与丙烯的摩尔比为1:1。
3)反应产物经过冷凝和气液分离后收集,收集到的产物进行gc检测后得甲醇的转化率为98.1%,主要产物包括1,3-丁二醇(选择性84.2%),4-甲基-1,3-二噁烷(选择性1.8%),四氢吡喃-4-醇(选择性3.1%)和二甲醚(选择性4.0%)。
实施例4
采用制备实施例4制备的催化剂进行试验。
1)在固定床反应器中加入10g催化剂4,在氢氮混合气(体积:5%h2-95%n2)下以3℃/min的升温速度升温至200℃还原3h后切换为氮气气氛,然后降温至100℃,压力6mpa。
2)用柱塞泵分别将丙烯(液体)和甲醇水溶液(90%)通入反应器中进行反应,以甲醇计反应空速为0.1h-1,甲醇与丙烯的摩尔比为1:1。
3)反应产物经过冷凝和气液分离后收集,收集到的产物进行gc检测后得甲醇的转化率为85.1%,主要产物包括1,3-丁二醇(选择性93.1%),4-甲基-1,3-二噁烷(选择性2.3%),四氢吡喃-4-醇(选择性2.1%)和二甲醚(选择性1.3%)。
图3为本实施例中反应产物的气相色谱图。图4为主要反应产物的质谱检测谱图。
实施例5
采用制备实施例5制备的催化剂进行试验。按照测试实施例1的方式合成1,3-丁二醇,结果显示甲醇的转化率为98.7%,主要产物包括1,3-丁二醇(选择性87.6%),4-甲基-1,3-二噁烷(选择性2.6%),四氢吡喃-4-醇(选择性4.4%)和二甲醚(选择性3.0%)。
实施例6
采用制备实施例6制备的催化剂进行试验。按照测试实施例1的方式合成1,3-丁二醇,结果显示甲醇的转化率为96.3%,主要产物包括1,3-丁二醇(选择性61.9%),二甲醚(选择性19.1%),4-甲基-1,3-二噁烷(选择性6.1%)和四氢吡喃-4-醇(选择性3.7%)。
实施例7
采用对比实施例1制备的对比催化剂进行试验。按照测试实施例1的方式合成1,3-丁二醇,结果显示甲醇的转化率为92.6%,主要产物包括二甲醚(选择性65.2%),1,3-丁二醇(选择性21.9%),4-甲基-1,3-二噁烷(选择性1.9%)和四氢吡喃-4-醇(选择性2.2%)。
由上述结果可知,催化剂制备实施例中所述催化剂对该反应有较高的反应活性与二元醇选择性,而对比实施例中所述催化剂虽然对甲醇有较高的反应活性,但是反应产物中二元醇的选择性较低。
- 还没有人留言评论。精彩留言会获得点赞!