一种大位阻氧杂螺环二酚及其双膦配体的合成方法与流程
1.本发明涉及一种大位阻氧杂螺环二酚骨架4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚,及其双膦配体的合成方法。
背景技术:
2.轴对称有机化合物一直是不对称催化领域的研究热点,轴对称化合物在生物医药、工业催化、功能材料等领域有着广泛的应用。已近成功工业化的binol、binap等联芳基配体已经有了很广泛的应用。
[0003][0004]
1992年,kumar等人将光学纯的cis,cis-螺[4,4]-1,6-壬二醇修饰的氢化铝锂,成功地应用于酮的不对称还原,取得了优秀的对映选择性(ee值最高可达98%)。这是首例关于手性螺环配体的催化性能研究的报道。1996年,keay等人将同样的手性二醇作为手性辅基修饰丙烯酰氯,成功地应用于环戊二烯的不对称diels-alder反应中。随后,陈新滋等人在此骨架基础上设计合成了不同类型的双齿亚膦酸酯配体,并研究了其在铑催化的不对称氢化和氢甲酰化反应中的应用。
[0005]
1999年,birman等从丙酮和3-甲氧基苯甲醛出发,经过六步反应得到了外消旋的螺二氢茚二酚((
±
)-spinol)。该二酚与氯甲酸薄荷醇酯形成的非对映异构体可以通过柱层析进行分离,从而得到光学纯的(r)-(+)-spinol和(s)-(-)-spinol。us20130135574a1、cn1055003542a也报道了类似的合成路线和拆分方法。在此基础上,南开大学的周其林等在2002年报道了更为实用的拆分方法,他们利用氯化苄基辛可宁啶和对映异构体中的一种易形成包结物的特点,通过简单的回流、冷却、结晶、过滤和酸化等步骤就可以得到光学纯的螺二氢茚二酚。2016年谭斌等人报道了手性膦酸催化下spinol的不对称合成,直接从1,5-双(5-羟基-2-甲基苯基)-3-戊酮一步环化脱水到(s)-4,4
’‑
二甲基-7,7
’‑
二羟基-1,1
’‑
螺二氢茚(收率97%,ee值90%)。值得一提的事,他们所使用的配体是以手性spinol为骨架的膦酸。此外,cn109761774a研究了一种从1,5-双(3-羟基苯基)-3-戊酮傅克环化到消旋spinol方法,这是首例有关羟基对位不需要占位基团即可环化合成1,1
’‑
螺二氢茚-7,7
’‑
二酚的报道。2018年,张绪穆等在螺二氢茚二酚的基础上,从1,3-二氟苯出发经六步反应得到了得到了外消旋的螺二氢苯并呋喃二酚((
±
)-o-spinol),该二酚可经l-脯氨酸拆分后得到光学纯的(r,s)-o-spinol。此外,cn109503659a、cn110128439a也报道了类似的合成路线和拆分方法。
[0006]
周其林小组基于螺环骨架的手性配体在不对称催化领域取得了巨大的成功,使螺环配体在短短数十年间发展成了一类重要的配体,他们的配体基本上都是基于螺二氢茚骨
架之上的。但是,和轴手性配体的发展相比,螺环骨架配体不仅是在数目上还是在应用上都显得较为不足,其中很重要的一个原因就是螺环骨架的合成比较困难。而且,以大位阻基团修饰spinol、o-spinol骨架以及合成其双膦配体报道的例子较少。因此发展新型大位阻氧杂螺环骨架(o-spinol)及其双膦配体具有深远的意义和非常高的研究价值。
[0007]
本发明中发展的一种新型大位阻氧杂螺环二酚及其双膦配体的方法具有易于合成、适合放大合成、可以产业化的工艺合成路线,该工艺路线简单,收率较高、所以原料均可回收利用等特点。此外,新型氧杂螺环双亚膦酸酯配体(消旋体)可用于催化烯烃的羰基化反应,该新型配体的手性化合物可用于催化不对称反应。
技术实现要素:
[0008]
本发明实施例的目的是在于提供一种大位阻氧杂螺环二酚及其双膦配体化合物。
[0009]
本发明的实施例是这样实现的,一种大位阻氧杂螺环二酚及其双膦配体化合物,其结构如通式i和ii所示:
[0010][0011]
其中,通式ii及其衍生物的结构表示如下:
[0012]
[0013][0014]
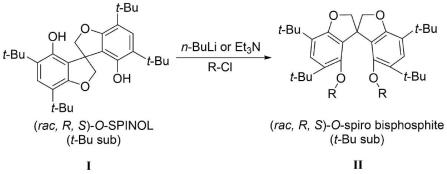
本发明实施例的另一目的在于提供一种大位阻氧杂螺环二酚及其双膦配体的合成方法,所述氧杂螺环双亚膦酸酯化合物是以4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚化合物为原料,在有机溶剂以及正丁基锂或三乙胺的作用下与含有芳基或环状芳基结构的氯代亚膦酸酯反应而成;所述氧杂螺环双亚膦酸酯配体为l1-l31中的一种。
附图说明
[0015]
图1,本发明配体化合物l4的1h nmr(600mhz,cdcl3)示意图;
[0016]
图2,本发明配体化合物l4的
31
pnmr(243mhz,cdcl3)示意图。
具体实施方式
[0017]
下面通过实施例对本发明的以上路线进行具体的描述,为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅用于解释本发明,并不用于限定本发明。
[0018]
本发明公开提供了合成路线,以1,3-二氟苯为起始原料,经锂化、亲核加成、脱水、羟醛缩合/坎尼扎罗(canizzaro)串联反应、两步芳香亲核取代(snar)和钯碳脱苄反应后得到消旋的氧杂螺二酚(o-spinol),再经烷基化反应得到四叔丁基取代大位阻的氧杂螺二酚,大位阻o-spinol消旋体可经拆分或手性催化后得到光学纯的大位阻氧杂螺二酚。消旋或光学纯的大位阻o-spinol可与氯代亚膦酸酯经酯化反应后得到氧杂螺环双亚膦酸酯配体。
[0019]
具体地说,本发明所涉及的合成方法具体说明如下:
[0020]
在一些实施方式中,1,3-二氟苯出发经正丁基锂锂化后,芳基锂试剂先与三甲基硅乙醇酸甲酯反应得到芳基酮,芳基酮再与芳基锂试剂发生亲核加成反应后,加入稀盐酸
水解同时脱掉tms保护基后得到1,1-双(2,6-二氟苯基)-1,2-乙二醇;
[0021]
在一些的实施方式中,所述起始原料除了1,3-二氟苯以外,也可以是1,3-二氯苯、1,3-二溴苯或1,3-二碘苯;优选1,3-二氟苯是因为在后续的snar反应中,氟是最好的离去基团。
[0022]
在一些的实施方式中,所述锂化反应中,锂试剂的用量为1~5当量,反应温度为-78~0℃,反应时间1~12小时,反应溶剂为乙醚、四氢呋喃、1,4-二氧六环、二氯甲烷等有机溶剂。
[0023]
在一些的实施方式中,所述亲核加成反应所用的有机锂类化合物为甲基锂、异丙基锂、正丁基锂、仲丁基锂、叔丁基锂或苯基锂中的任意一种。
[0024]
在一些实施方式中,1,1-双(2,6-二氟苯基)-1,2-乙二醇在硫酸中加热回流后脱水得到1,1-双(2,6-二氟苯基)-乙醛。
[0025]
在一些实施方式中,1,1-双(2,6-二氟苯基)-乙醛在碱催化下与多聚甲醛先发生羟醛缩合反应得到1,1-双(2,6-二氟苯基)-3-羟基丙醛,再发生坎尼扎罗反应(歧化反应)得到1,1-双(2,6-二氟苯基)-1,3-丙二醇。
[0026]
在一些实施方式中,所述羟醛缩合/坎尼扎罗反应所用的碱为氢氧化钠、氢氧化钾、氢氧化锂、甲醇钠、乙醇钠、叔丁醇钠、叔丁醇钾中的任意一种。反应溶剂为乙醚、四氢呋喃、1,4-二氧六环、甲基叔丁基醚中的任意一种。碱的用量为5~80当量,反应温度为50~120℃,反应时间为6~12个小时。
[0027]
在一些实施方式中,1,1-双(2,6-二氟苯基)-1,3-丙二醇上的羟基与离去基团在缚酸剂的作用下发生芳香亲核取代(snar)反应,环化后得到1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二氟。
[0028]
在一些实施方式中,所述snar反应所用的缚酸剂可以为有机碱或无机碱中的一种或多种,有机碱如::三乙胺,n,n-二异丙基乙胺,吡啶等;无机碱如:碳酸铯、碳酸钾、碳酸锂、叔丁醇钠、叔丁醇钾、氢化钠、氢氧化钠或氢氧化钾等;缚酸剂用量为5~100当量,反应温度为-10~80℃,反应时间为2~10个小时;反应溶剂为乙醚、四氢呋喃、1,4-二氧六环、甲基叔丁基醚等有机溶剂。
[0029]
在一些实施方式中,1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二氟与苯甲醇在缚酸剂的作用下发生芳香亲核取代(snar)反应,得到1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二苄醚。
[0030]
在一些实施方式中,所述snar反应所用的缚酸剂为碳酸铯、碳酸钾、碳酸锂、叔丁醇钠、叔丁醇钾中的任意一种;缚酸剂用量为2~40当量,反应温度为25~140℃,反应溶剂为甲苯、对甲苯、对二甲苯、邻二甲苯、氯苯、二氯苯或dmf中的任意一种。
[0031]
在一些实施方式中,1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二苄醚在钯碳的作用下发生脱苄反应,得到消旋的1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(o-spinol)。
[0032]
在一些实施方式中,所述脱苄反应中的金属催化剂钯碳的含量为5%或以上,催化剂的用量为5~10%(w/w),氢气压力1~10mpa,反应温度为25~40℃,反应时间为5~12个小时;反应溶剂为乙醚、四氢呋喃、1,4-二氧六环、甲基叔丁基醚等有机溶剂。
[0033]
在一些实施方式中,在质子酸或路易斯酸的催化下,叔丁醇脱水后生成异丁烯,异丁烯与o-spinol发生亲电加成反应得到4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚。
[0034]
在一些实施方式中,所述烷基化反应所用的质子酸或路易斯酸为有机酸或无机酸中的一种或多种,有机酸如:甲酸、乙酸、乙二酸、二氯乙酸、三氟乙酸、丙酸、丙二酸、丙酮酸、丁酸、戊酸、己酸、己二酸、苯甲酸、对硝基苯甲酸、对苯二甲酸、苯磺酸、氟磺酸、甲磺酸、三氟甲磺酸、对甲苯磺酸等;无机酸如:氢溴酸、盐酸、氢氟酸、亚硫酸、硫酸、高氯酸、膦酸、焦磷酸、硝酸、亚硝酸、铬酸、氟锑磺酸、氟锑酸等;烷基化试剂为溴代叔丁烷、氯代叔丁烷、异丁烯、叔丁醇中的任意一种;反应温度为50~110℃,反应溶剂为苯、甲苯、对甲苯、对二甲苯、邻二甲苯、氯苯或二氯苯中的任意一种。
[0035]
在一些实施方式中,4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚与正丁基锂反应得到锂化后反应液;锂化后反应液与含有芳基或环状芳基结构的氯代亚膦酸酯反应,得到大位阻氧杂螺环双亚膦酸酯化合物。
[0036]
在一些实施方式中,4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚与含有芳基或环状芳基结构的氯代亚膦酸酯和缚酸剂的混合溶液反应,得到大位阻氧杂螺环双亚膦酸酯化合物。
[0037]
在一些实施方式中,所述酯化反应中,正丁基锂的用量2~4当量,缚酸剂为三乙胺、n,n-二异丙基乙胺、吡啶中的任意一种,用量为5~20当量,反应温度为-78~80℃,反应时间12~48小时,反应溶剂为甲苯、四氢呋喃、乙醚、2-甲基四氢呋喃、甲基叔丁基醚、异丙醚、苯甲醚、乙二醇二甲醚、二乙二醇二甲醚、丁醚、环戊基甲醚或1,4-二氧六环中的任意一种。
[0038]
实施例1:1,1-双(2,6-二氟苯基)-1,2-乙二醇的制备(3)
[0039][0040]
在一个干燥的1l的schlenk瓶加入1(50.0g,437mmol)和300ml无水四氢呋喃,将反应瓶置换为氮气氛围,待反应液冷却至-78℃后缓慢滴加2.5m的正丁基锂的己烷溶液(176ml,1.005eq.)。滴加完毕后,在-78℃下搅拌1小时,然后缓慢加入三甲基硅乙醇酸甲酯(34.77g,214mmol)。滴加完毕后,反应升温至-30℃继续搅拌8小时后升至室温。反应结束后,在-20℃下加入稀盐酸淬灭反应,同时脱去三甲基硅基保护基。反应液用乙醚和二氯甲烷萃取,合并有机相,减压除去溶剂后得到目标产物41.9g,收率65%。1hnmr(400mhz,cdcl3):δ=3.54
–
3.70(m,1h),4.55(d,2h),4.73(s,1h),6.96(m,4h),7.44(m,2h)。
[0041]
实施例2:1,1-双(2,6-二氟苯基)-乙醛的制备(4)
[0042][0043]
在1l的双口瓶加入3(20.0g,69.9mmol)和110ml质量分数为26%的硫酸,100℃下回流反应4小时。冷却至室温后,有机相用二氯甲烷萃取,无水硫酸钠干燥。减压旋干得到粗产物,通过快速柱层析分离后得到目标产物17.1g,收率92%。1h nmr(400mhz,cdcl3):δ=
5.30(s,1h),6.72
–
6.85(m,4h),7.12
–
7.20(m,2h),9.84(m,1h)。
[0044]
实施例3:1,1-双(2,6-二氟苯基)-1,3-丙二醇的制备(6)
[0045][0046]
在500ml的双口瓶加入4(13.4g,50mmol)、氢氧化锂(24.0g,1000mmol),多聚甲醛(30.0g,1000mmol)和120ml无水二氧六环。将反应瓶置换为氮气氛围,80℃下反应10小时。加入稀盐酸淬灭反应,混合液用乙醚和二氯甲烷萃取,合并有机相,减压旋干后得到粗品,通过快速柱层析分离后得到目标产物13.5g,收率90%。1h nmr(400mhz,cdcl3):δ=2.34(br,2h),4.52(s,4h),6.75-6.79(m,4h),7.10-7.20(m,2h)。
[0047]
实施例4:1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二氟的制备(7)
[0048][0049]
在250ml的双口瓶加入6(5.0g,16.7mmol)和叔丁醇钾(5.6g,50.0mmol),将反应瓶置换为氮气氛围后于0℃下加入80ml无水四氢呋喃,然后将反应瓶恢复至室温后,在60℃下反应7小时,加入稀盐酸淬灭反应,二氯甲烷萃取有机相,无水硫酸钠干燥,减压旋干后得到目标产物4.2g,收率97%。1hnmr(400mhz,cdcl3):δ=4.64(d,2h),4.79(d,2h),6.57-6.61(m,2h),6.68(d,2h),7.15-7.20(m,2h)。
[0050]
实施例5:1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二苄醚的制备(8)
[0051][0052]
在200ml的双口瓶加入7(4.0g,15.4mmol)和叔丁醇钾(10.3g,61.5mmol),将反应瓶置换为氮气氛围后,加入苯甲醇(6.6g,61.5mmol)和100ml无水n,n-二甲基甲酰胺。在100℃下反应8小时,冷却反应体系至室温,加入大量的水后析出白色固体,过滤即得目标产物6.6g,收率99%。1hnmr(400mhz,cdcl3):δ=4.50(d,2h),4.78(d,2h),4.80(d,2h),4.86(d,2h),6.39(d,2h),6.43(dd,2h),6.78-6.80(m,4h),7.06-7.09(m,8h)。
[0053]
实施例6:1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(o-spinol)的制备((rac)-9)
[0054][0055]
在100ml的高压反应釜内,依次加入8(6.0g,13.8mmol),300mg催化剂(10%pd/c)
和50ml四氢呋喃。往釜内充入4mpa氢气后室温反应20小时,减压旋干得到粗品,并通过快速柱层析分离得到白色固体产物3.5g,收率99%。1h nmr(400mhz,dmso):δ=4.50(d,2h),4.58(d,2h),6.23-6.27(m,4h),6.92(dd,2h),6.78-6.80(m,4h),7.06-7.09(m,8h)。
[0056]
实施例7:4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚的制备(rac-o-spinol-t-bu)
[0057][0058]
在200ml的三口瓶中依次加入(rac)-9(3.0g,11.7mmol)、叔丁醇(5.5g,74.2mmol)、浓硫酸(3.6g,37.1mmol)。加料完毕后,将反应瓶置换为氮气氛围,加热至回流反应24小时。将溶剂减压旋干,加入50ml水,用乙酸乙酯萃取有机相,无水硫酸钠干燥后减压旋干,剩余物经快速柱层析即得5.4g目标产物,收率96%。1h nmr(400mhz,cdcl3):δ=1.40(d,36h),4.53(d,2h),4.69(d,2h),6.75(s,2h),7.14(s,2h)。
[0059]
实施例8:1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(o-spinol)的拆分
[0060][0061]
在250ml圆底瓶中,依次加入rac-9(12.8g,50mmol)、l-脯氨酸(2.9g,25mmol),然后加入100ml乙酸乙酯。在80℃下搅拌8小时,期间有白色固体析出,冷却至室温过滤并收集白色固体。将白色固体加入到乙酸乙酯和水的混合溶剂中超声,固体逐渐溶解消失。用乙酸乙酯萃取有机相,无水硫酸钠干燥后减压旋干,乙酸乙酯中重结晶后得到光学纯的(s)-9(ee大于99%)。用同样的方法就可以得到光学纯的(r)-9(ee大于99%)。
[0062]
实施例9:7,7
’‑
二[(1,1
’‑
联苯-2,2
’‑
二基)亚膦酸酯]-4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃的制备(l4)
[0063][0064]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(4.0g,8.3mmol),无水三乙胺(17.3ml,124.8mmol,
15.0eq.)和无水四氢呋喃100ml。然后,将混合物冷却至-30℃后滴加1,1
’‑
联苯-2,2
’‑
二氧基氯化膦(4.8g,19.1mmol,2.3equiv.)的50ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,粗品快速柱层析分离后,用乙腈重结晶得到目标产物5.1g,收率68%。1h nmr(600mhz,cdcl3):δ=1.18-1.24(d,36h),4.75(d,2h),4.96(d,2h),6.91
–
7.23(m,10h),7.27
–
7.30(m,4h),7.41
–
7.44(m,4h);
31
pnmr(243mhz,cdcl3):δ=145.18。
[0065]
实施例10:不对称氧杂螺环双亚膦酸酯配体的制备(rac-l43)
[0066][0067]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(4.0g,8.3mmol),无水三乙胺(8.7ml,62.4mmol,7.5eq.)和无水四氢呋喃100ml。然后,将混合物冷却至-20℃后滴加3,3’,5,5
’‑
四叔丁基-1,1
’‑
联苯-2,2
’‑
二氧基氯化膦(4.7g,10.0mmol,1.2equiv.)的70ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,粗品快速柱层析分离后,用乙腈重结晶得到目标产物rac-l42(6.3g,收率82%)。
[0068]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入rac-l42(6.3g,6.9mmol),无水四氢呋喃100ml,在-20℃下滴加入2.5m正丁基锂(2.8ml,6.9mmol,1.0eq.)。反应混合物升至室温后回流反应1小时。然后-40℃下将该反应液滴入2-氯-1,3,2-苯并二氧膦杂环己烷-4-酮(1.68g,8.3mmol,1.2equiv.)的20ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,残余物柱层析获得目标产物rac-l43(3.8g,收率51%)。1h nmr(600mhz,cdcl3):δ=1.34
–
1.38(m,36h),1.45
–
1.46(m,36h),4.45
–
4.75(m,4h),7.03
–
7.47(m,9h),7.93(dd,1h);
31
pnmr(243mhz,cdcl3):δ=123.41,140.36。
[0069]
实施例11:(r,r)-7,7
’‑
二[(1,1
’‑
联苯-2,2
’‑
二基)亚膦酸酯]-4,4’,6,6
’‑
四叔丁基-(r)-1,1
’‑
螺二氢苯并呋喃的制备((r,r,r)-l17)
[0070][0071]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入(r)构型的4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢茚-7,7
’‑
二酚(4.0g,8.3mmol),无水三乙胺(17.3ml,124.8mmol,15.0eq.)和无水四氢呋喃100ml。然后,将混合物冷却至-40℃后滴加(r)-(1,1
’‑
联萘-2,2’‑
二氧基)氯化膦(7.6g,21.6mmol,2.6equiv.)的100ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,粗品快速柱层析分离后,用乙腈重结晶得到目标产物(r,r,r)-l17(9.2g,收率72%)。1h nmr(600mhz,cdcl3):δ=1.37
–
1.45(d,36h),4.52
–
4.66(dd,4h),7.05(s,2h),7.30
–
7.43(m,16h),7.87
–
7.94(m,8h);
31
p nmr(243mhz,cdcl3):δ=143.08。
[0072]
实施例12:2,2
’‑
双[(二吡咯基膦基)氧代]-4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃的制备(l32)
[0073][0074]
在一个干燥的500ml的schlenk瓶中于氮气保护下依次加入4,4’,6,6
’‑
四叔丁基-1,1
’‑
螺二氢苯并呋喃-7,7
’‑
二酚(4.0g,8.3mmol),无水三乙胺(17.3ml,124.8mmol,15.0eq.)和无水四氢呋喃100ml。然后,将混合物冷却至-30℃后滴加1,1'-(氯膦二基)双(1h-吡咯)(4.0g,19.9mmol,2.4equiv.)的50ml无水四氢呋喃溶液中,滴完后室温反应24小时,氮气氛围下将反应液浓缩,粗品快速柱层析分离后,用乙腈重结晶得到目标产物5.1g,收率77%。1h nmr(600mhz,cdcl3):δ=1.45(d,36h),4.45
–
4.73(dd,4h),6.25(t,8h),6.91(t,8h),7.21(s,2h);
31
pnmr(243mhz,cdcl3):δ=125.93。
[0075]
这里要指出的是,通式ii中其它的l1-l41的氧杂螺环双膦配体,只需要使用不同的氯代亚膦酸酯或亚膦酰胺取代基衍生物即可制备得到。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1