一种碘介导α-重氮酯氧化快速生成α-酮酯的制备方法
本发明涉及酮酯类化合物研究领域,具体为一种碘介导α-重氮酯氧化快速生成α-酮酯的方法。
背景技术:
α-酮酯是一种带有相邻羰基和酯基两种官能团的化合物,不仅广泛存在于自然界中,而且能够表现出各种类型的生物活性。它们在医药化学和材料化学中被广泛应用为合成众多杂环结构的中间体。例如:在nh4oac存在下,苯甲酸苄酯与醛缩合,容易得到在生物活性药物中具有重要作用的氮杂环咪唑(yarabhally,r.g.;kuntebommanahalli,n.t.;kanchugarakoppal,s.r.,etal.rsc.adv.2015,5:75533-75546)。以α-酮酯,巯基乙酸碳离子缩合合成含硫杂环化合物-多取代噻吩(traversone,a.;brill,w.k.-d.de.tetrahedronlett.2007,48:3535-3537)。此外,moineau-chaneching还证明了除了合成有机杂环外,α-酮酯还可以快速合成具有光学性质的异感镍配合物(thanh-tuan,b.;alix,s.-s.;alain,m.,etal.inorg.chem.2014,53:2841-2847)。由于α-酮酯邻近的二羰基,所以也被用作许多有价值配体的前体,如1,2-二醇(deluca,l.;mezzetti,a.angew.chem.int.ed.2017,56:11949-11953),1,2-二亚胺(guo,l.;gao,h.;guan,q.,etal.organometallics.2012,31:6054-6062),nhcs(hongbo,l.;pratt,r.c.;culkin,d.a.,etal.chem.commun.2006,2881-2883)等。α-酮酯更是治疗胃和十二指肠溃疡的药物-安胃灵的中间体(陈韶,陈克明,吕海云,廖艾梅,张菊生.北京师院学报(自然科学版).1986,01:27-30),治疗前列腺癌药物-比卡鲁胺的中间体(夏泉,孟玲,范文玉.精细与专用化学品.2006,14(6):14-17)和用作杀菌剂-噻菌灵的中间体(孔繁蕾,缪留福,吴仲芳.江苏化工.1988,4:32-33)。在合成α-酮酯的方法中,α-重氮酯的氧化脱氮是一种有用的方法。典型的氧化策略一般是用过硫酸氢钾和丙酮生成的4,5-二甲基-1,3-二氧杂环戊烯-2-酮环丙烷(dmdo)((a)ming,m.;changkun,l.;lingling,p.,etal.tetrahedronlett.2005,46:3927-3929.(b)truong,p.;shanahan,c.s.;doyle,m.p.org.lett.2012,14:3608-3611.(c)rubin,m.b.;gleiter,r.chem.rev.2000,100:1121-1164)或者叔丁基次氯酸(tbuocl)作为强氧化剂来氧化α-重氮酸酯(roβbach,j.;baumeister,j.;harms,k.;koert,u.eur.j.org.chem.2013,2013:662-665)。但是这些方法反应条件苛刻,强氧化的使用容易造成大规模生产安全隐患,并且底物局限性很大。
2006年hu等报道了醋酸铑二聚体催化α-重氮酸酯和水反应生成α-酮酯,需要加入难以降解的偶氮二甲酸二乙酯(guo,z.;huang,h.;fu,q.;hu,w.synlett.2006,15:2486-2488)。此外,stoltz和他的同事发现二甲基亚砜也可以作为氧化剂实现α-重氮酸酯的脱氮氧化反应,但他们的底物仅局限于具有供电子基团的重氮酸酯(o’connor,n.r.;bolgar,p.,etal.tetrahedronlett.2016,57:849-851)。
2015年jeong,j.等利用吡啶-n-氧化物作为氧化剂实现了α-重氮酸酯的氧化,但需要贵金属铑作为催化剂且产率适中(yu,y.;sha,q.;cui,h.,etal.org.lett.2018,20:776-779)。
2020年changmingxu等利用单质氧作为氧化剂在cui催化下实现了α-重氮酸酯的氧化为α-酮酯(cheng,m.,x.;yang,c.w.;lei,b.org.chem.2020,85:12579-12584),该方法也需要较高的温度并且产率也未有显著的提升。
可见,对于这类反应的改进仍然存在的很大的挑战。本发明就是针对上述方法的缺点进行研究,首次探索出一种条件温和、环境友好、底物范围广且有实用性的高效制备α-酮酯的普适方法。
技术实现要素:
本发明的目的是提供一种高效、反应条件温和、环境友好的制备α-酮酸酯的方法。
一种碘介导α-重氮酯氧化快速生成α-酮酯的制备方法,其制备方法的反应式表示如下:
其中,式中的r1是指氢、c1-30烷基、c6-14芳基、c4-14杂芳基、c2-30烯基、c3-30炔基或者卤素;r2是指c1-20烷基、c6-14芳基、c4-14杂芳基、c2-30烯基、c3-30炔基;即在有机溶剂中,采用α-重氮酯和碘单质为原料,在室温下搅拌至少半小时,即可制得α-酮酯化合物。
进一步的,所述碘与α-重氮酯的摩尔比是1:1~3:1。
进一步的,所述有机溶剂包括四氢呋喃、乙腈、甲苯、二氯甲烷其中之一。
进一步的,所述α-酮酸酯化合物的产率为80%~99%。
与现有技术相比,本发明的有益效果是:
本发明是目前已知的合成α-酮酯的最理想的方法,其优点在于以下几个方面:(1)反应温度为室温,反应时间短且高效,为半小时左右。(2)本发明无需使用过渡金属作为催化剂,也无需使用强氧化剂或者共氧化剂。(3)本发明是环境友好、底物范围广的高效制备α-酮酯的普适方法。
附图说明
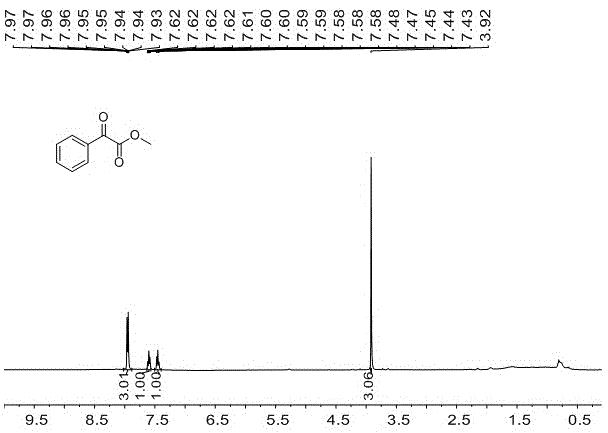
图1是本发明实施例1的氢谱(产率94%);
图2是本发明实施例2的氢谱(产率99%);
图3是本发明实施例3的氢谱(产率86%);
图4是本发明实施例4的氢谱(产率80%);
图5是本发明实施例5的氢谱(产率90%);
图6是本发明实施例6的氢谱(产率87%);
图7是本发明实施例7的核磁粗谱。
具体实施方式
通过下述实施例将有助于理解本发明,但并不限制本发明的内容。下面结合附图对本发明实施例作进一步说明:
一种碘介导α-重氮酯氧化快速生成α-酮酯的制备方法可以用下述典型的反应式来表示:
其中,式中的r1是指氢、c1-30烷基、c6-14芳基、c4-14杂芳基、c2-30烯基、c3-30炔基或者卤素;r2是指c1-20烷基、c6-14芳基、c4-14杂芳基、c2-30烯基、c3-30炔基。
具体的说就是在包括四氢呋喃、乙腈、甲苯或者二氯甲烷其中之一的有机溶剂中,采用的碘单质和α-重氮酯(摩尔比为1:1~3:1)为原料,室温搅拌半小时左右,可以制得α-酮酯化合物。在整个制备过程中无需使用过渡金属作为催化剂,也无需使用强氧化剂或者共氧化剂。
α-酮酯的制备步骤:在干燥的10ml支口管中,将α-酮酯化合物(0.2mmol,1equiv.)溶于四氢呋喃(2ml)中,加入i2(0.2mmol,1equiv.),在室温下搅拌。薄层色谱跟踪直至反应结束,约30分钟。将得到的混合物加入饱和硫代硫酸钠水溶液,并用乙酸乙酯萃取三次,无水硫酸镁干燥,浓缩,并通过硅胶色谱柱纯化,得到α-酮酯化合物。
实施例1:本发明通过上述制备步骤,由α-苯基-α-重氮乙酸甲酯(0.20mmol,35.2mg)合成。薄层色谱跟踪反应(乙酸乙酯:石油醚=1:10,rf=0.5)。快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到图1所示(0.189mmol,30.8mg,产率为94%)。1hnmr(400mhz,cdcl3)δ8.02–7.88(m,3h),7.63–7.57(m,1h),7.45(t,j=7.6hz,1h),3.92(s,3h).。
实施例2:α-(4-苯基苯基)-α-酮乙酸甲酯:根据上述制备步骤,由α-(4-苯基苯基)-α-重氮乙酸甲酯(0.20mmol,50.2mg)合成。薄层色谱跟踪反应(乙酸乙酯:石醚=1:10,rf=0.5)。快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到如图2所示(0.19mmol,47.5mg,产率99%)。1hnmr(400mhz,cdcl3)δ8.10(d,j=8.6hz,2h),7.72(d,j=8.6hz,2h),7.65–7.60(m,2h),7.48(ddt,j=8.1,6.5,1.1hz,2h),7.45–7.39(m,1h),4.00(s,3h)。
实施例3:α-(4-氟苯基)-α-酮乙酸甲酯:根据上述制备步骤,由α-(4-氟苯基)-α-重氮乙酸甲酯(0.20mmol,38.8mg)合成。薄层色谱跟踪反应(乙酸乙酯:石油醚=1:10,rf=0.5)。快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到如图3所示(0.18mmol,31.3mg,产率86%)。1hnmr(400mhz,cdcl3)δ8.13–8.01(m,2h),7.17(t,j=8.6hz,2h),3.96(s,3h)。
实施例4:α-(4-氰基苯基)-α-酮乙酸甲酯:根据上述制备步骤,由α-(4-氰基苯基)-α-重氮乙酸甲酯(0.20mmol,40.2mg)合成。薄层色谱跟踪反应(乙酸乙酯:石油醚=1:10,rf=0.4)。快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到如图4所示(0.16mmol,30.3mg,产率80%)。1hnmr(400mhz,cdcl3)δ8.15(d,j=8.7hz,2h),7.81(d,j=8.7hz,2h),3.99(s,3h)。
实施例5:α-苯基-α-酮乙酸环戍酯:根据上述制备步骤,由α-苯基-α-重氮乙酸环戍酯(0.20mmol,40.6mg)合成。薄层色谱跟踪反应(乙酸乙酯:石油醚=1:10,rf=0.7)快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到如图5所示(0.18mmol,39.2mg,产率90%)。1hnmr(400mhz,cdcl3)δ7.93–7.86(m,2h),7.56(dd,j=8.4,6.5hz,1h),7.42(t,j=7.8hz,2h),5.40(tt,j=6.0,2.9hz,1h),1.95–1.86(m,2h),1.83–1.75(m,2h),1.72–1.65(m,2h),1.59–1.54(m,2h)。
实施例6:α-苯基-α-酮乙酸-2-戍炔酯:根据上述制备步骤,由α-苯基-α-重氮乙酸-2-戍炔酯(0.20mmol,45.6mg)合成。薄层色谱跟踪反应(乙酸乙酯:石油醚=1:10,rf=0.7)快速层析色谱分离(洗脱剂:乙酸乙酯:石油醚=1:30)得到如图6所示(0.17mmol,37.6mg,产率87%)。1hnmr(400mhz,cdcl3)δ8.02(d,j=7.7hz,2h),7.66(t,j=7.5hz,1h),7.51(t,j=7.7hz,2h),4.95(t,j=2.3hz,2h),2.26(qt,j=7.5,2.2hz,2h),1.16(t,j=7.5hz,3h)。
实施例7:以0.2mmolα-苯基-α-重氮乙酸甲酯(35.2mg)和0.2mmol的碘单质(51mg)为反应模型,在室温空气的条件下反应直至薄层色谱监测反应结束。反应完成后将得到的混合物加入30ml饱和硫代硫酸钠水溶液,并用乙酸乙酯萃取三次,合并有机层,无水硫酸镁干燥,浓缩,得到α-酮酯化合物粗产,在双排管上抽干溶剂,加入0.2mmol的内标物ch2br214μl,氘代氯仿3ml,通过核磁共振氢谱400hz1hnmr测试产物的含量。
如图7所示,内标物ch2br2中的-ch2化学位移为4.84,产物α-苯基-α-酮乙酸甲酯中的-och3化学位移为3.89。通过氢个数比例计算产率,可以算得2.86÷3≈95。所以从核磁粗谱可以看出,产率为95%。
以上的展示的仅为本发明实施例而已,当然不能以此来限定本发明的权利范围,以此依靠本发明权利要求所作的同等变化,仍属于本发明所涵盖的范围。
- 还没有人留言评论。精彩留言会获得点赞!