一种1,3,4-噁二唑衍生物及其制备方法和用途
一种1,3,4
‑
噁二唑衍生物及其制备方法和用途
技术领域
1.本发明公开了一种1,3,4
‑
噁二唑衍生物,公开了这种1,3,4
‑
噁二唑衍生物的不同的制备方法和其新的用途。其能够用于制备共济失调毛细血管扩张突变基因和rad3相关激酶(atr)的抑制剂和抗肿瘤药物。
背景技术:
2.共济失调毛细血管扩张突变基因和rad3相关激酶(ataxia telangiectasia and rad
‑
3related protein kinase,atr)作为pikk家族中的重要蛋白激酶,不仅和基因重组有关,而且在dna损伤修复通路中发挥关键调节作用。atr接受上游调控信号单链dna(single stranded dna,ssdna)的断裂被激活后可通过磷酸化一系列下游蛋白,控制细胞周期分裂,促进脱氧核糖核苷酸合成,从而确保基因组的完整性。atr的生理学功能受损可导致一系列疾病,包括免疫缺陷、神经系统紊乱和癌症。由于肿瘤细胞中dna损伤应答机制存在缺陷,表现出肿瘤细胞对atr依赖性更高,atr抑制剂可以选择性抑制肿瘤细胞而对健康细胞影响较小。因此,atr被视为理想的抗肿瘤药物靶点。
3.1,3,4
‑
噁二唑及其衍生物作为一种重要的医药中间体,具有光谱生物活性,在消炎、抗真菌、抗结核、抗hiv、抗病毒以及抗肿瘤等方面广泛应用。传统的1,3,4
‑
噁二唑衍生物的制备方法中最常用的有“双酰肼环合法”,该合成方法通常需要较高的回流温度和脱水剂(如三氯氧磷、五氯氧磷、浓硫酸、亚硫酰氯、三氟甲磺酰酰氯和多聚磷酸等),上述脱水剂存在毒性强、沸点高、腐蚀性高且对环境污染严重等问题,不利于实验室使用和工业化生产。尽管采用微波辅助反应方法快捷高效,但缺点是安全系数低。本发明以二溴三苯基膦为脱水环化剂,室温条件即可发生反应,收率高、反应后处理简单并且对环境友好,具有很好的实用价值。
4.现有的atr抑制剂数量较少,只有两种小分子药物(vx
‑
970和azd6738)处于临床试验阶段,迄今并没有一款上市药物的开发。渥曼青霉素(wortmannin)和五味子乙素(schisandrin b)是早期开发的两种弱选择性的atr抑制剂,因其药理毒性限制了进一步研究。诺华公司开发的一款吡唑并哌嗪衍生物对atr的抑制活性为ic
50
=0.4nm,由于其存在体内药物代谢的安全性问题,该衍生物主要用作化学增敏剂使用。az20是一款开发的低纳摩尔级别活性的atr抑制剂,其抑制atr的活性为ic
50
=5nm,可以使移植lovo结直肠细胞的小鼠肿瘤体积显著减小,但该抑制剂的低溶解度限制了进一步研究。因此,针对现阶段atr抑制剂数量不足、溶解度低且存在临床试验不良副反应等问题,开发新型的特异性atr抑制剂仍具有重要实际意义。
技术实现要素:
5.本发明属于药物研究领域,涉及一种1,3,4
‑
噁二唑衍生物,公开其与往不同的制备方法,以及其与往不同的用途,用途包括用于制备共济失调毛细血管扩张突变基因和rad3相关激酶(atr)的抑制剂和抗肿瘤药物。
6.本发明公开的一种1,3,4
‑
噁二唑衍生物,其具有如下结构:
[0007][0008]
本发明提供了所述1,3,4
‑
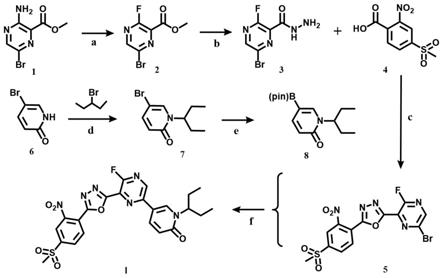
噁二唑衍生物的制备方法,通过图1的化学合成路线制备。其中,该制备方法具体包含以下步骤:
[0009]
步骤a:以化合物1(3
‑
氨基
‑6‑
溴吡嗪
‑2‑
甲酸甲酯)为原料,加入盐酸的水溶液使其酸化。冷却反应体系,滴加亚硝酸钠(nano2)以及体积百分含量为40%的四氟硼酸(hbf4)水溶液,化合物1、盐酸、亚硝酸钠以及四氟硼酸的摩尔比为1:2.5:1.2:3,反应混合物在室温反应3h后减压抽滤,得到化合物1的中间体(3
‑
四氟硼酸铵
‑6‑
溴吡嗪
‑2‑
甲酸甲酯)为淡黄色粉末状物质。将3
‑
四氟硼酸铵
‑6‑
溴吡嗪
‑2‑
甲酸甲酯溶解于甲苯中,反应混合物于110℃回流1h,使3
‑
四氟硼酸铵
‑6‑
溴吡嗪
‑2‑
甲酸甲酯充分分解。反应完毕真空减压浓缩除去甲苯,粗产物经过硅胶柱分离纯化得到化合物2(3
‑
氟
‑6‑
溴吡嗪
‑2‑
甲酸甲酯)。
[0010]
步骤b:以化合物2(3
‑
氟
‑6‑
溴吡嗪
‑2‑
甲酸甲酯)为底物,以无水乙醇为溶剂,加入水合肼进行酯交换反应,反应混合物于70℃回流反应1.5h,化合物2和水合肼的摩尔比为1:6,回流反应后反应混合物冷却至室温,减压过滤并用无水乙醇洗涤滤饼,得到化合物3(6
‑
溴
‑3‑
氟吡嗪
‑2‑
碳酰肼)。
[0011]
步骤c:以化合物3(3
‑
氟
‑6‑
溴吡嗪
‑2‑
碳酰肼)和化合物4(2
‑
硝基
‑4‑
(甲基磺酰基)苯甲酸)为底物,以无水乙腈为溶剂,以二溴三苯基膦为脱水环化剂,室温搅拌反应1小时,滴加n,n
‑
二异丙基乙胺(diea)继续反应16h。化合物3、化合物4、二溴三苯基膦和n,n
‑
二异丙基乙胺的摩尔比为1:1:3:6。反应后处理为减压过滤并用无水乙腈和正己烷洗涤滤饼,得到化合物5(2
‑
(6
‑
溴
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑)。
[0012]
步骤d:以化合物6(5
‑
溴
‑
2(1h)
‑
吡啶酮)和3
‑
溴戊烷为底物、以乙二醇二甲醚为溶剂,加入3
‑
溴戊烷和碳酸铯,化合物6、3
‑
溴戊烷和碳酸铯的摩尔比为1:1.5:3,反应混合物于80℃回流2h。反应后处理为用乙酸乙酯和饱和食盐水萃取,洗涤后合并有机层,用无水na2so4干燥并过滤,真空减压浓缩,粗产物经过硅胶柱分离纯化得到化合物7(5
‑
溴
‑1‑
异戊基
‑
2(1h)
‑
吡啶酮)。
[0013]
步骤e:以化合物7(5
‑
溴
‑1‑
异戊基
‑
2(1h)
‑
吡啶酮)和联硼酸频哪醇酯为底物,以1,4
‑
二氧六环为溶剂,以醋酸钾和二氯化钯为催化剂;化合物7、联硼酸频哪醇酯、醋酸钾以及二氯化钯的摩尔比为1:1.5:2:0.1,氮气保护条件下反应混合物于90℃回流12h后用乙酸乙酯和饱和食盐水洗涤,洗涤后合并有机层,用无水na2so4干燥,过滤并浓缩,粗产物经过硅胶柱分离纯化得到化合物8(1
‑
异戊基
‑6‑
氧代
‑
1,6
‑
二氢吡啶
‑3‑
硼酸频哪醇酯)。
[0014]
步骤f:以化合物5(2
‑
(6
‑
溴
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑)和化合物8(1
‑
异戊基
‑6‑
氧代
‑
1,6
‑
二氢吡啶
‑3‑
硼酸频哪醇酯)为底物,以1,4
‑
二氧六环为溶剂,以1,1'
‑
双二苯基膦二茂铁二氯化钯为催化剂,加入2m碳酸钾水溶液。化合物5、化合物8、1,1'
‑
双二苯基膦二茂铁二氯化钯和碳酸钾的摩尔比为1:1.1:0.1:
2,氮气保护条件下反应混合物于80℃回流反应2小时。回流反应后处理为用饱和食盐水洗涤,洗涤后合并有机层,用无水na2so4干燥并过滤,真空减压浓缩,粗产物经过硅胶柱分离纯化得到化合物ⅰ(2
‑
(6
‑
(1
‑
异戊基
‑
2(1h)
‑
吡啶酮
‑4‑
基)
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑)。
[0015]
本发明所述的1,3,4
‑
噁二唑衍生物结构新颖,侧链苯环的吸电子基团(硝基)有助于提升其抗肿瘤活性。该1,3,4
‑
噁二唑衍生物的制备方法与传统1,3,4
‑
噁二唑衍生物制备方法(如“双酰肼环合法”)相比,能克服高温反应条件、脱水环化剂毒性强且易腐蚀等缺点,本发明提供的制备方法采用二溴三苯基膦为脱水环化剂,室温即可反应且对环境友好。本发明公开的1,3,4
‑
噁二唑衍生物用途与以往用途(如抗菌、抗病毒等)不同,可用于atr抑制剂和协同抗乳腺癌药物的制备。
附图说明
[0016]
图1是本发明1,3,4
‑
噁二唑衍生物的制备路线
[0017]
图2是本发明1,3,4
‑
噁二唑衍生物的1h nmr谱图
[0018]
图3是本发明1,3,4
‑
噁二唑衍生物的
13
c nmr谱图
[0019]
图4是本发明1,3,4
‑
噁二唑衍生物的ms谱图
[0020]
图5是本发明盐酸阿霉素抑制mcf
‑
7细胞的增殖活性
[0021]
图6是本发明1,3,4
‑
噁二唑衍生物抑制mcf
‑
7细胞的增殖活性
[0022]
图7是本发明1,3,4
‑
噁二唑衍生物与盐酸阿霉素联合给药抑制mcf
‑
7细胞的增殖活性
具体实施方式
[0023]
以下结合附图,通过具体的实施例对本发明作进一步描述,这些实施例仅用于说明本发明,并不是对本发明保护范围的限制。
[0024]
化合物ⅰ制备的具体实施方法如下:
[0025]
步骤a:将3
‑
氨基
‑6‑
溴吡嗪
‑2‑
甲酸甲酯(5.0g,21.55mmol)溶于盐酸(5.47ml,53.88mmol)的水溶液(盐酸与水的体积比为1:1)中搅拌至糊状,室温反应20分钟。将该反应体系冷却至0℃后,滴加20ml nano2(1.64g,23.7mmol)水溶液,在0℃下充分搅拌反应10min后,滴加体积比为40%的hbf4水溶液(14.2ml,66.45mmol),滴加完毕后在室温继续反应3小时。反应混合物减压抽滤,滤饼用冰水洗涤,真空干燥箱干燥,得淡黄色粉末状物质3
‑
四氟硼酸铵盐
‑6‑
溴吡嗪
‑2‑
甲酸甲酯。将上述3
‑
四氟硼酸铵盐
‑6‑
溴吡嗪
‑2‑
甲酸甲酯(4.5mg,19.1mmol)溶解于20ml甲苯中,充分搅拌反应混合物于110℃回流1h,使氟硼酸重氮盐充分分解,同时利用naoh溶液吸收分解产生的酸性气体bf3,防止其对环境产生污染。真空减压浓缩除去甲苯溶液,通过硅胶柱分离纯化得到化合物2(3
‑
氟
‑6‑
溴吡嗪
‑2‑
甲酸甲酯),收率为20%。
[0026]
步骤b:将3
‑
氟
‑6‑
溴吡嗪
‑2‑
甲酸甲酯(800mg,3.42mmol)溶解于30ml无水乙醇中,滴加水合肼(85%,0.75ml,20.5mmol),滴加完毕于70℃回流反应1.5h。反应液在冰箱冷却结晶,析出大量橙黄色沉淀,减压过滤得6
‑
溴
‑3‑
氟吡嗪
‑2‑
碳酰肼,收率为85%。
[0027]
步骤c:将6
‑
溴
‑3‑
氟吡嗪
‑2‑
碳酰肼(500mg,1.2mmol)溶于无水乙腈(5ml),分别加
入2
‑
硝基
‑4‑
(甲基磺酰基)苯甲酸(314mg,1.44mmol),二溴三苯基膦(1522mg,3.6mmol)室温搅拌反应1小时后,滴加n,n
‑
二异丙基乙胺(1.672ml,9.6mmol)。反应混合物室温搅拌16小时。用饱和食盐水洗涤,洗涤后合并有机层,用na2so4干燥,过滤并浓缩,粗产物经过硅胶柱分离纯化得到2
‑
(6
‑
溴
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑,收率为32.5%。
[0028]
步骤d:将5
‑
溴
‑
2(1h)
‑
吡啶酮(1.159mg,6.66mmol)、碳酸铯(2.82g,8.7mmol)溶于乙二醇二甲醚溶液(20ml)中,室温滴加3
‑
溴戊烷(8.64ml,8.7mmol)。滴加完毕,反应混合物于80℃回流反应2小时。用乙酸乙酯和饱和盐水洗涤,洗涤后合并有机层,用无水na2so4干燥并过滤,真空减压浓缩,粗产物经过硅胶柱分离纯化得到中间体5
‑
溴
‑1‑
异戊基
‑
2(1h)
‑
吡啶酮,收率为69.5%。
[0029]
步骤e:将5
‑
溴
‑1‑
异戊基
‑
2(1h)
‑
吡啶酮(1.0g,4.63mmol)溶于1,4
‑
二氧六环(15ml)中,依次加入联硼酸频那醇酯(1.41g,5.55mmol),醋酸钾(0.61g,9.3mmol)以及二氯化钯(339mg,0.462mmol)。氮气保护条件下反应混合物于90℃回流反应12小时。用乙酸乙酯和饱和食盐水洗涤,洗涤后合并有机层,用无水na2so4干燥,过滤并浓缩,粗产物经过硅胶柱分离纯化得到黑色油状液体(1
‑
异戊基
‑6‑
氧代
‑
1,6
‑
二氢吡啶
‑3‑
硼酸频哪醇酯),收率为82.5%。
[0030]
步骤f:将2
‑
(6
‑
溴
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑(200mg,0.5mmol)溶于1,4
‑
二氧六环(20ml),分别加入1
‑
异戊基
‑6‑
氧代
‑
1,6
‑
二氢吡啶
‑3‑
硼酸频那醇酯(161mg,0.55mmol),1,1'
‑
双二苯基膦二茂铁二氯化钯(36.8mg,0.05mmol)以及2m碳酸钾(0.504ml,1.0mmol)。氮气保护条件下反应混合物于80℃回流反应2小时。用饱和食盐水洗涤,洗涤后合并有机层,用na2so4干燥,过滤并浓缩,粗产物经过硅胶柱分离纯化得到最终化合物ⅰ为2
‑
(6
‑
(1
‑
异戊基
‑
2(1h)
‑
吡啶酮
‑4‑
基)
‑3‑
氟吡嗪
‑2‑
基)
‑5‑
(2
‑
硝基
‑4‑
(甲基磺酰基)苯基)
‑
1,3,4
‑
噁二唑,收率为58.2%。该化合物ⅰ经过核磁共振氢谱表征其结构(图2):1h nmr(400mhz,c2d6so)δ:0.83(t,6h),1.63(m,2h),1.69(m,2h)3.12(s,3h),4.67(m,1h),6.35(d,1h),7.44(d,1h),7.66(s,1h),8.12(d,2h),8.32(d,2h)8.41(s,1h),8.53(s,1h)。该化合物ⅰ经过核磁共振碳谱表征其结构(图3):
13
c nmr(400mhz,c2d6so)δ:10.35,21.00,24.59,42.95,52.30,83.71,119.00,122.86,131.17,131.65,142.20,144.31,150.07,154.96,162.34,165.37,166.10,171.95。该化合物ⅰ经质谱分析(图4),ms(es
+
)m/z:584.85[m+k+h2o]
+
。
[0031]
本发明使用上述1,3,4
‑
噁二唑衍生物抑制mcf
‑
7细胞增殖的实验考察其抗肿瘤用途。本发明使用的细胞株为人源乳腺癌细胞mcf
‑
7,来源于中国科学院上海细胞库,阳性对照药物为盐酸阿霉素(doxorubicin hydrochloride,dox)。实验采用cck
‑
8试剂盒检测细胞生长情况,根据细胞存活率计算ic
50
,进而评价新型atr抑制剂的抗肿瘤效果。
[0032]
本发明实验分为3组,分别为给药组(分别为化合物单独给药组及化合物与盐酸阿霉素联合给药组)、阴性对照组(加入体积分数0.1%的无菌dmso完全培养基溶液)和空白对照组。设置新型atr抑制剂的浓度梯度为25μm、50μm、100μm、200μm和400μm;盐酸阿霉素的浓度梯度为0.01μm、0.1μm、1μm、10μm和100μm。盐酸阿霉素与新型atr抑制剂以摩尔比1:1联合使用的浓度梯度为0.02μm、0.2μm、2μm、20μm以及200μm。
[0033]
本发明抗mcf
‑
7细胞增殖实验的具体实施过程如下:
[0034]
a.细胞培养:人源乳腺癌细胞株(mcf
‑
7)用dmem完全培养基(含有10%体积分数的胎牛血清和1%体积分数的青链霉素(双抗))在37℃、5%co2恒温培养箱培养24~48h,显微镜下观察mcf
‑
7细胞的生长状态,待细胞贴壁生长后从培养箱取出。
[0035]
b.细胞接种:取对数生长期的mcf
‑
7细胞,弃掉原有培养基,用pbs缓冲液清洗细胞,加入1ml胰酶消化液消化2min。然后加入2ml dmem完全培养基终止消化。将经胰酶消化的细胞转移至15ml离心管中,放入离心机于1500rpm离心5min,弃去上清液之后用dmem完全培养基重悬细胞,细胞混匀后接种于96孔板中,每孔100μl。之后将接种细胞的96孔板放回恒温培养箱中继续培养24h,显微镜定期观察细胞至贴壁生长。
[0036]
c.给药处理:用无菌dmso分别配制化合物ⅰ和盐酸阿霉素最大浓度母液,再用dmem完全培养基分别按比例稀释成不同浓度梯度的溶液。按照浓度梯度每孔加入100μl配制的候选化合物和盐酸阿霉素溶液,每个浓度设置3个复孔,将96孔板放进培养箱培养24~48h。
[0037]
d.cck
‑
8检测法计算细胞存活率:用完全培养基配制含10%体积分数的cck
‑
8检测试剂,弃去每孔中原有的培养基,加入pbs缓冲液清洗细胞后加入cck
‑
8检测试剂,放回恒温培养箱继续培养1~2h,最后用酶标仪在450nm波长处读取od值,按照公式1计算细胞存活率。按照公式2计算药物协同作用指数(combination index,ci),从而判断两种药物的协同效应。
[0038][0039][0040]
其中,a、b代表不同的两种药物,d
a
和d
b
分别代表两种药物联合使用使肿瘤细胞生长抑制率达到x时的药物浓度,ic
x,a
和ic
x,b
分别代表两种药物单独使用使肿瘤细胞生长抑制率达到x时的药物浓度。
[0041]
本发明公开的一种1,3,4
‑
噁二唑衍生物对mcf
‑
7细胞具有增殖抑制作用。根据公式1计算得盐酸阿霉素抑制mcf
‑
7细胞的增殖活性为ic
50
=0.854μm(图5),1,3,4
‑
噁二唑衍生物抑制mcf
‑
7细胞增殖活性为ic
50
=154.087μm(图6)。
[0042]
盐酸阿霉素与1,3,4
‑
噁二唑衍生物以摩尔比1:1联合使用抑制mcf
‑
7细胞增殖活性为0.597μm(图7),根据公式2计算得其协同效应ci=0.351。根据soriano等的联合用药指数法:当0.9≤ci≤1.1时,药物发挥相互拮抗作用;当0.8≤ci<0.9时,药物发挥低度协同作用;当0.6≤ci<0.8时,药物发挥中度协同作用;当0.4≤ci<0.6时,药物发挥高度协同作用;当0.2≤ci<0.4时,药物发挥强协同作用。表明本发明提供的一种1,3,4
‑
噁二唑衍生物与盐酸阿霉素联合使用抑制mcf
‑
7细胞增殖发挥强协同作用,能够显著抑制mcf
‑
7肿瘤细胞增殖。
[0043]
本发明提供的一种1,3,4
‑
噁二唑衍生物,其用途主要为抑制mcf
‑
7肿瘤细胞增殖,采用化疗药物盐酸阿霉素与新型atr抑制剂协同抗肿瘤增殖的方案,能够减少盐酸阿霉素的用量,以期降低化疗药物长期使用产生的耐药性问题,该策略有望为肿瘤临床治疗提供新的思路和解决方案。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1