一种利用AIE分子识别和拆分手性化合物的方法
本发明属于手性分子技术领域,涉及一种手性化合物的识别和拆分技术,具体涉及一种利用aie分子识别和拆分手性化合物的方法。
背景技术:
传统的荧光生色团在高浓度下荧光会减弱甚至不发光,这种现象被称作“浓度猝灭”效应。浓度猝灭的主要原因跟聚集体的形成有关,故浓度猝灭效应通常也被叫做“聚集导致荧光猝灭(aggregation-causedquenching,acq)”。
2001年,唐本忠课题组发现了一个奇特的现象:一些噻咯分子在溶液中几乎不发光,而在聚集状态或固体薄膜下发光大大增强,因为此发光增强是由聚集所导致的,故我们形象地将此现象定义为“聚集诱导发光(aggregation-inducedemission,aie)”。
手性分子是指与其镜像不相同不能互相重合的具有一定构型或构象的分子,20世纪中期的“反应停事件”就是一个突出的例子。研究发现,消旋体“反应停”中,有镇静作用的是它的(r)-异构体,而(s)-异构体具有致畸作用。惨痛的教训使人们认识到,手性药物必须对它的两个异构体进行分别检验,慎重对待。一些药物的另一对映异构体,表现有不良作用的例子还很多。
目前已经有一定的手性分子识别方法,例如色谱法和传感器法,但是成本昂贵、难以操作。因此急需开发一种低成本的手性化合物快速鉴别方法。
技术实现要素:
本发明的目的在于建立一种操作简单、快速、低成本的识别和拆分手性化合物的方法。
荧光法是一种操作简单的高灵敏度检测法,基于此方法,本发明人合成了一种手性aie分子,该分子与手性酸在溶剂中络合后的溶解度降低进而呈现aie效应,通过对络合后的荧光光谱光谱进行分析,识别手性化合物。而且可以利用手性酸与aie分子络合后的状态不同(如:溶液与沉淀)实现对手性化合物的拆分,由此得到了本发明。
综上所述,本发明提供了一种aie分子,所述aie分子具有如下r/s-7或r/s-10所述的化学结构:
其中,取代基r1为烷基;取代基r2选自卤素、氰基、三氟甲基、烷基中的一种。
进一步地,本发明所述的烷基为具有通式cnh2n+1(n为大于0的整数)的正烷基、异烷基、新烷基。优选的,所述烷基选自甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基中的一种。
进一步地,所述卤素选自cl、br、i中的一种。
本发明人对上述r/s-7或r/s-10进行了研究,探究出了一种合成方法。具体的,所述r/s-7通过以下路径进行制备:
,所述r/s-10通过以下路径进行制备:
本发明还提供了所述aie分子在手性化合物识别中的应用,具体的所述识别手性化合物的方法是:
分别配置aie分子溶液和手性化合物溶液;
将手性化合物溶液与aie分子溶液混匀;
静置络合;
根据荧光检测结果区分手性化合物的类别。
本发明所述通过aie分子识别手性化合物的方法,其原理是aie分子与手性化合物络合后会产生不同的荧光强度,可根据其荧光强度的差异来区分手性化合物的类别。
作为本发明优选的实施方式,本发明所述的aie分子优选的用于手性酸的识别,所述手性酸包括l型或d型的boc-谷氨酸。
进一步地,配置aie分子溶液和手性化合物的溶液时,溶剂优选的为1,2-二氯乙烷。
进一步地,手性化合物与aie分子的摩尔量之比等于手性化合物分子上羧基与aie分子上的氨基个数之比。
另外,aie分子和手性化合物络合后,若络合作用较强,会生成类盐的物质,从而析出。而作用较弱的组仍会以溶液状态存在,就可以通过抽滤把作用较强的手性酸给分离出来。基于此,本发明人提供了一种利用aie分子拆分手性化合物的方法,具体包括如下的方法或者步骤:
配置aie分子溶液和手性化合物的溶液;
将手性化合物溶液与aie分子溶液混匀;
静置络合;
过滤分离手性化合物。
本发明提供的aie分子在手性化合物识别和拆分中的应用。
需要说明的是,本发明提供的aie分子是手性的,但做识别时,只用到其中的单一手性aie分子,去识别或者拆分一对手性酸,另一个aie分子做识别和拆分时会表现出正好相反的结果。例如,r-7与d型的boc-谷氨酸作用后荧光增强,与l型的boc-谷氨酸作用后荧光几乎不变。而s-7就会与l型的boc-谷氨酸作用后荧光增强,与d型的boc-谷氨酸作用后无荧光变化,这是由于手性空间构型相反造成的,两个手性分子可以互相参照。
经本发明人试验研究,本发明提供的aie分子优选的用于手性酸的识别与拆分,但是并非对本发明提供的aie分子的使用范围作出限定,也有可能用于醇类手性化合物的识别与拆分。因此,本发明对所述aie分子的使用范围不做限定,任何在不脱离本发明设计精神的前提下,利用本发明提供的aie分子或类似结构的分子用于任何手性化合物识别与拆分,均应落入本发明确定的保护范围。
本发明与现有技术相比,具有以下有益效果或者优点:
本发明提供了一种利用aie分子识别和拆分手性化合物的方法,其原理是利用本发明提供的手性aie分子与手性化合物络合后在溶剂中状态的差异,来实现手性化合物的识别与拆分。相对于传统的hplc等分离手性化合物的方法,本发明利用荧光可视化识别与拆分手性酸的方法操作起来更为简单,成本更低。
附图说明
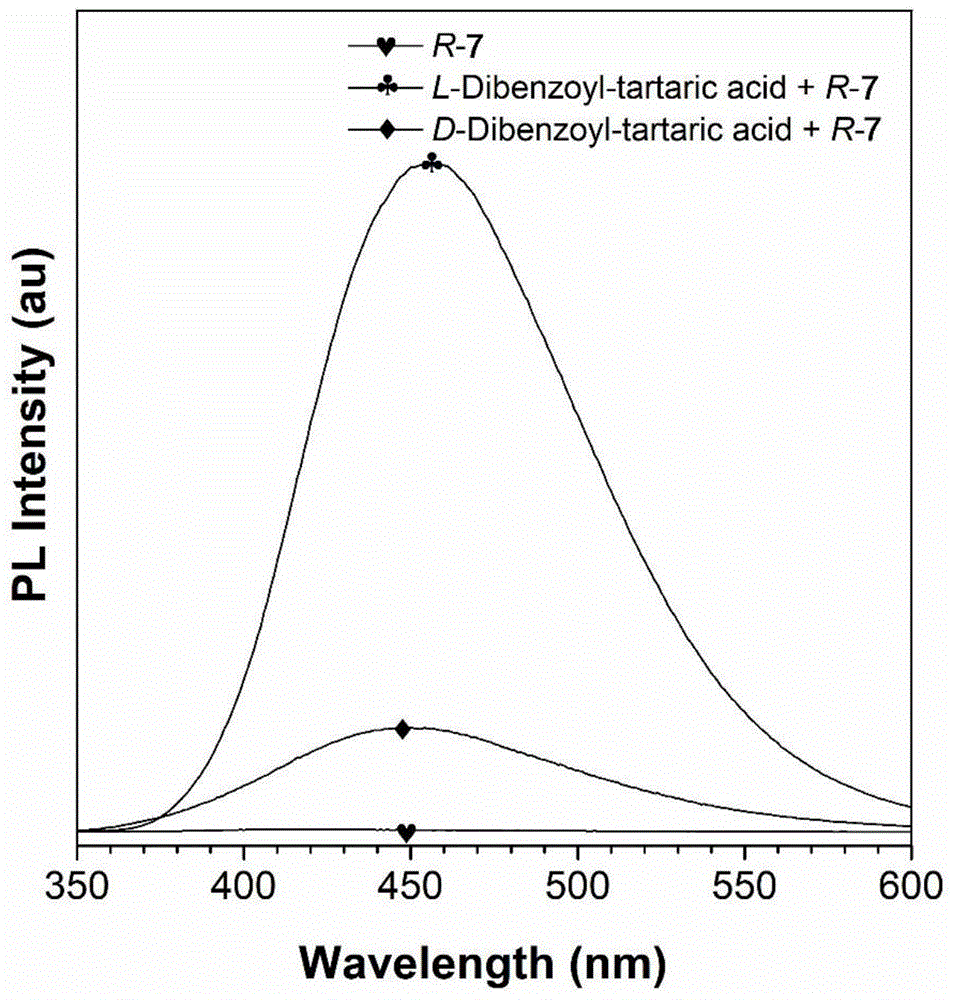
图1为[r-7]=[l-二苯甲酰基-酒石酸]=[d-二苯甲酰基-酒石酸]=(5.0×10-4m)在添加正己烷的1,2-二氯乙烷中的荧光光谱变化图(条件:λex=325nm,λex/λem狭缝=5/5nm)。
图2为三种溶液在365nm紫外灯照射下的照片。
图3为在日光灯(a)和便携式365nm紫外线灯(b)下,2mgr-7与boc-谷氨酸在1.0ml1,2-二氯乙烷中形成的凝胶的照片。图a中,左为boc-d-谷氨酸+r-7,右为boc-l-谷氨酸+r-7。
具体实施方式
下面,结合附图对本发明的技术方案进行进一步的解释说明,但是,本发明并不限于下述的实施方式。
为了建立一种简单、快速识别和拆分手性化合物的方法,本发明提供了一种aie分子,用于手性化合物的识别和拆分。具体的,所述aie分子具有如下r/s-7或r/s-10的结构:
其中,取代基r1为烷基;取代基r2选自卤素、氰基、三氟甲基、烷基中的一种。
所述烷基选自甲基、乙基、丙基、丁基、戊基、己基、庚基、辛基中的一种。
所述卤素选自cl、br、i中的一种。
其中,r/s-7通过以下方法或者步骤进行制备:
具体的,制备方法如下:
化合物2的合成:取250ml的三颈圆底烧瓶加入二苯基甲烷(5.8g,34.68mmol),然后抽真空3min,再缓慢通入氮气2min,如此交换循环3次后,最后在氮气保护下,注入70ml新蒸四氢呋喃,开启搅拌,将反应液冰浴至0℃,然后用恒压滴液漏斗逐滴滴入20.60ml正丁基锂(33.03mmol,2.5mol/l)溶液,0.5h滴加完毕,0℃条件下继续搅拌0.5h后用固体加料漏斗加入化合物1(8.0g,33.0mmol),撤去冰浴,室温反应过夜。待原料反应完全之后逐滴滴加60ml饱和nh4cl水溶液淬灭反应,分出有机相,水层用三氯甲烷萃取三次后与有机相合并,加入无水mgso4干燥,然后减压过滤mgso4固体,旋转蒸发得白色固体,再用chcl3-ch3oh重结晶得白色固体12.3g,收率90.7%,此反应中产率均以r1=r2=h为例。
化合物3的合成:取250ml的两颈圆底烧瓶,将化合物2(12.3g,33.03mmol)和对甲基苯磺酸(1.3g,6.60mmol)加入其中,再注入150ml甲苯,装上分水器和回流冷凝管,加热回流脱水2.5h,用tlc监测反应进程,待原料2反应完全后,将反应液冷却至室温,有机相用饱和nahco3水溶液洗涤三次,接着水相用chcl3萃取两次后与有机相合并,加入无水mgso4干燥,减压过滤,旋转蒸发除去溶剂得到白色固体,再用chcl3-ch3oh重结晶得白色固体11.9g,22收率91.1%,此反应中产率均以r1=r2=h为例。
化合物4的合成:取250ml的圆底烧瓶,加入化合物3(10.0g,25.51mmol)和120ml二氯甲烷,用液n2-乙醇混合液将反应体系冷却至-40℃,然后将bbr3(6.6ml,71.40mmol)溶于30ml二氯甲烷中,通过恒压滴液漏斗缓慢滴加进反应体系,滴加完后继续低温反应10min,然后室温反应8h,用tlc监测反应进程,反应结束后加入80ml冰水淬灭反应,析出大量白色沉淀,分出有机层,水相用乙酸乙酯萃取三次,合并有机相,加入无水mgso4干燥,过滤,减压蒸发除去溶剂得到8.8g白色固体,收率95.0%,此反应中产率均以r1=r2=h为例。
化合物6的合成:取50ml的两颈圆底烧瓶,将化合物5(244mg,0.58mmol)和化合物6(152mg,1.28mmol)加入其中,化合物6为单一手性r型和s型,需做两次反应;使之溶解于20ml无水乙醇中,升温回流,4h后,用tlc监测反应过程。反应结束后,直接抽滤,烘干;得到白色粉末,r型和s型的产率分别为87%和89%,此反应中产率均以r1=r2=h为例。
化合物r/s-7的合成:取100ml的两颈圆底烧瓶,将化合物6(150mg,0.235mmol)加入其中,使之溶解于20ml无水乙醇/四氢呋喃(1:1)中;在冰浴下,30分钟内缓慢分三次加入硼氢化钠(88mg,2.35mmol),然后撤去冰浴,室温反应;12h后,用tlc监测反应过程。反应结束后,停止反应;旋干溶剂,用乙酸乙酯和水萃取三次,合并有机相,加入无水mgso4干燥,过滤,减压蒸发除去溶剂后,再用chcl3-hexane重结晶得白色固体,r型和s型的产率分别为84.8%和76.2%,此反应中产率均以r1=r2=h为例。
r/s-10通过以下方法或者步骤进行制备:
具体的,制备方法如下:
化合物9的合成:取50ml的两颈圆底烧瓶,将化合物4(252mg,0.599mmol)和化合物8(160mg,1.319mmol)加入其中,化合物8为单一手性r型和s型,需做两次反应;使之溶解于20ml无水乙醇中,升温回流,4h后,用tlc监测反应过程。反应结束后,直接抽滤,烘干;得到白色粉末,r型和s型的产率分别为94%和96%,此反应中产率均以r1=r2=h为例。
化合物r/s-10的合成:取100ml的两颈圆底烧瓶,将化合物9(150mg,0.239mmol)加入其中,使之溶解于20ml无水乙醇/四氢呋喃(1:1)中;在冰浴下,30分钟内缓慢分三次加入硼氢化钠(135mg,3.5897mmol),然后撤去冰浴,室温反应;12h后,用tlc监测反应过程。反应结束后,停止反应;旋干溶剂,用乙酸乙酯和水萃取三次,合并有机相,加入无水mgso4干燥,过滤,减压蒸发除去溶剂后,r型和s型的产率分别为94%和98.7%,此反应中产率均以r1=r2=h为例。
荧光识别中化合物r-7在1,2-二氯乙烷中配置成5×10-4m的液,取3组3ml的待测,然后分别配置两瓶5×10-2m手性酸(如:l型和d型的boc-谷氨酸)的1,2-二氯乙烷溶液,从两瓶手性酸溶液中取30ul分别加入两组待测的r-7溶液中,命名为l组和d组,未加手性酸溶液的待测r-7溶液为空白组。等待络合5min后,在325nm的波长激发下,测试其350-600nm范围内的发射波长,并观测其络合后的状态,得到表1所示的结果:
表1,化合物r-7对不同手性化合物的识别作用
表中,pre表示沉淀物;sus表示浮液;sol表示溶液。dce=1,2-二氯乙烷;运用的检测探针为r-7。
由表1可知,以r-7作为手性酸的探针,可实现对多种手性酸的识别作用;例如:r-7与不同手性的n-叔丁基氧羰基-谷氨酸作用后,荧光强度可以达到103倍的差异。并且r-7与n-叔丁基氧羰基-d-谷氨酸作用后可生成凝胶,与n-叔丁基氧羰基-l-谷氨酸没有作用,仍然呈现溶液状态。r-7与不同手性的二苯甲酰基-酒石酸作用后,也可以有4.5倍的荧光差异。
如图1和图2所示,r-7与不同手性的二苯甲酰基-酒石酸作用后均在450nm处有荧光发射峰,未产生明显的峰位置移动。说明此处为r-7形成络合物之后的发射峰,而且由于r-7与l-二苯甲酰基-酒石酸作用更加强烈,形成的络合物溶解性更差,因此产生的荧光也更强。图2在365nm紫外灯下的照片更能明显的反映出荧光的差异。
如图3所示,r-7与n-叔丁基氧羰基-d-谷氨酸作用后可生成凝胶,从图片a中可以看出r-7与n-叔丁基氧羰基-d-谷氨酸作用后,由于两者作用力较强,在溶液中络合后会呈现出明显的凝胶状态,而与n-叔丁基氧羰基-l-谷氨酸作用未有明显的作用。图片b在紫外灯下可以观测到荧光的明显差异,生成凝胶后的荧光强度明显高于溶液状态。根据这种络合后的不同状态就可以实现对手性酸的拆分。
如上所述,即可较好地实现本发明,上述的实施例仅仅是对本发明的优选实施方式进行描述,并非对本发明的范围进行限定,在不脱离本发明设计精神的前提下,本领域普通技术人员对本发明的技术方案做出的各种改变和改进,均应落入本发明确定的保护范围内。
- 还没有人留言评论。精彩留言会获得点赞!