伏立康唑的合成工艺的制作方法
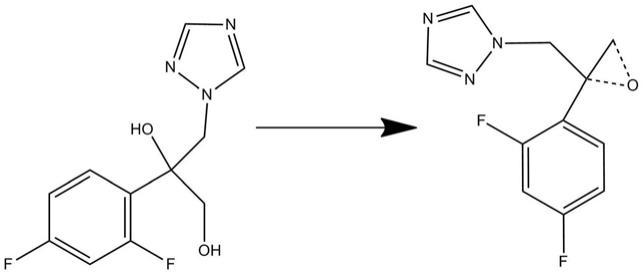
1.本发明涉及药物合成领域,尤其是涉及伏立康唑原料药的合成工艺,特别涉及一种伏立康唑及其消旋体的制备。
背景技术:
2.伏立康唑(voriconazole,vrc,uk109496),其化学名为(2r,3s)
‑2‑
(2,4
‑
二氟苯基)
‑3‑
(5
‑
氟嘧啶-4
‑
基)
‑1‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑2‑
丁醇,是美国辉瑞公司在氟康唑基础上合成的一种新型抗真菌药,主要用于进行性、有致命危险的免疫损害患者。由于伏立康睡抗真菌谱广、抗菌效力强,安全性好,且国内市场对抗真菌药物的需求增长迅速,因此市场前景巨大。
3.现有的合成伏立康唑的方法或工艺一般是以2r,3s/2s,3r消旋体为关键中间体,使用特定的手性拆分试剂,如1r
‑
(
‑
)
‑
10
‑
樟脑磺酸,成盐拆分得到伏立康唑。然而,传统上用于制备伏立康唑或其中间体的方法工艺一般存在收率低、产物纯度不高等问题。
4.因此,亟需一种操作简便、工艺可控、收率高、纯度高、适宜工业化生产的伏立康唑原料药的合成工艺,由此制备满足需求的伏立康唑及其消旋体。
技术实现要素:
5.针对现有技术中存在的上述问题,本发明提供了伏立康唑原料药或其消旋体的合成方法工艺,以特定的卤代烷基
‑
氟代嘧啶和二氟苯基
‑
三唑基丙二醇类化合物为原料,在离子液体存在下进行制备,从而合成得到伏立康唑消旋体,本发明提供的方法工艺反应条件温和,无需引入大量助剂和溶剂,并且分离纯化简单,能够以较高收率获得伏立康唑及其消旋体。
6.为了实现上述目的,第一方面,本发明提供了一种伏立康唑消旋体的制备方法,其包括:
7.步骤1、准备卤代乙基氟代嘧啶,并进行格氏反应;
8.步骤2、将2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇进行氧化,得到环氧丙烷化合物;
9.步骤3、将步骤1所得的格氏试剂和环氧丙烷化合物混合,进行反应,得到伏立康唑。
10.第二方面,本发明提供了一种伏立康唑或其消旋体,其由第一方面的工艺合成制备。
11.本发明伏立康唑的合成工艺能够取得以下有益效果:
12.(1)本发明通过利用2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇作为反应原料,直接生成伏立康唑消旋体,能够简化反应步骤,无需使用钯碳进行脱氯氢解,缩短反应周期,能够节约能耗、降低成本,从而能够提高伏立康唑消旋体的收率;
13.(2)本发明后处理为洗涤,过滤,其操作简便且无需萃取等复杂工艺;
14.(3)本发明的伏立康唑消旋体可直接用于制备伏立康唑,无需进一步纯化,从而节约物料成本和时间成本,且所得伏立康唑纯度高,满足药物质量标准;
15.(4)本发明操作简单、收率高、工艺安全性高、无需引入大量助剂和溶剂,具有节能、环保等优点,适合工业化和产业化。
附图说明
16.图1是本发明实施例2伏立康唑的质谱图。
具体实施方式
17.下面对本发明的较佳实施例进行详细阐述,以使本发明的优点和特征能更易于被本领域技术人员理解,从而对本发明的保护范围做出更为清楚明确的界定。
18.根据本发明第一方面,提供伏立康唑的合成工艺,该工艺方法包括以下步骤:
19.步骤1、准备卤代乙基氟代嘧啶,并进行格氏反应。
20.在本发明中,所述卤代乙基氟代嘧啶优选为4
‑
(1
‑
卤代乙基)
‑5‑
氟嘧啶,卤代基可以为cl
‑
、br
‑
、i
‑
中的任一者,优选为br
‑
。该4
‑
(1
‑
溴乙基)
‑5‑
氟嘧啶可以市购,如湖北永阔科技有限公司或湖北齐飞医药化工有限公司,也可以由价格便宜、容易获得的4
‑
乙基
‑
5氟嘧啶与常用的溴化剂,如n
‑
溴代丁二酰亚胺(nbs)反应得到。
21.根据本发明,4
‑
(1
‑
卤代乙基)
‑5‑
氟嘧啶与金属镁进行格氏反应,得到卤化镁格氏试剂,反应过程如下:
[0022][0023]
在本发明的优选实施方式中,在步骤1中,所用的金属镁为镁屑。
[0024]
本发明人发现,镁的选择比较重要。由于镁粉会使反应剧烈,或者镁粉表面容易形成氧化膜阻碍反应的进行,且镁粉不容易纯化。因此,本发明选择镁屑作为原料。
[0025]
本发明优选的实施方式中,镁屑在进行反应之前,需要预处理,包括:先将镁屑用稀酸洗刷,抽滤;然后用丙酮洗涤,真空干燥,铝箔包裹,置于n2或惰性气体下保存。
[0026]
在本发明中,步骤1中使用的反应溶剂i为醚类和/或叔胺类,优选地,醚类选自丙醚、丁醚、戊醚、异丙醚、异戊醚、苯甲醚、苯乙醚、甲基叔丁基醚、四氢呋喃和甲基四氢呋喃中的至少一者,叔胺类选自n,n
‑
二甲苯胺和/或吡啶。更优选地,反应溶剂i选择为四氢呋喃和/或甲基四氢呋喃,例如为甲基四氢呋喃。
[0027]
其中,甲基四氢呋喃的沸点高(80℃),能够提高反应速度,且生成的副产物较少,同时降低溶剂冷凝回收时的损失。甲基四氢呋喃在水中的溶解性比四氢呋喃低,因此更容易地将格氏试剂独立地包含在其中,同时回收干燥的甲基四氢呋喃更容易。另外甲基四氢呋喃有着更清晰的分相能力,因此本发明优选选择甲基四氢呋喃做反应溶剂i。
[0028]
四氢呋喃的沸点为66℃,其做溶剂时可能产生副产物联苄,但是反应容易引发。在反应中,可以先用四氢呋喃引发反应后在补加甲基四氢呋喃。
[0029]
其中,在反应之前,反应溶剂i可以先进行预处理。该预处理过程为向反应溶剂i中加入koh干燥3
‑
5天,然后蒸馏得到无水的反应溶剂i。
[0030]
在本发明的优选实施方式中,4
‑
(1
‑
卤代乙基)
‑5‑
氟嘧啶和反应溶剂i的摩尔比为1:(8~20),优选为1:(10~16),更优选为1:15。
[0031]
根据本发明,当4
‑
(1
‑
卤代乙基)
‑5‑
氟嘧啶和反应溶剂i的摩尔比为1:(8~20)时,控制其滴加量,能够减少副产物联苄的产生。
[0032]
其中,反应温度对制备格氏试剂至关重要。若温度过高,则会发生偶联反应,若温度过低,可能不引发。因此,优选地,反应温度为35~60℃,优选为45~55℃,例如50℃。
[0033]
根据本发明,当反应温度为45~55℃时,反应能够达到最佳。
[0034]
其中,微量的水分就能够抑制反应的引发,而且会分解形成的格氏试剂而影响产率。同时,若反应容器中未采取氮气或惰性气体置换,反应容器内溶剂蒸发产生大量易燃气体,与空气混合形成爆炸性混合气体,容易爆炸。因此,在反应进行前,向反应容器中通入氮气或惰性气体,如氦气、氖气、氩气等,以除去反应容器中的空气,使得反应在氮气或惰性气体气氛下进行。
[0035]
示例性地,为了缩短引发时间,在步骤1中,还可以包括加入少量的引发剂。优选地,所述引发剂选自碘或二氯甲烷,例如加入1
‑
2粒碘。
[0036]
步骤2、将2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇进行氧化,得到环氧丙烷化合物。
[0037]
在本发明中,所述2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇可以市购,如广州牌牌生物科技有限公司或美国qcc公司,也可以根据文献keiji tamura etal.the journal of organic chemistry,2014,79(7),3272
‑
3278的方法制得。
[0038]
该步骤反应过程为:
[0039][0040]
在本发明的优选实施方式中,在步骤2中,所用催化剂为负载型催化剂,其中催化剂载体选自氧化铝、氧化硅、活性炭和沸石分子筛中的至少一者;负载物质为碱金属可溶性盐或者碱金属氧化物,优选选自氧化锂、氧化钠和氧化钾的至少一者。
[0041]
其中,不同载体和不同负载物质制备负载型催化剂的方法相类似。示例性的,负载型催化剂可以按以下过程进行制备:
[0042]
(1)制备浸渍液,制备一定浓度的碱金属可溶性盐或者碱金属氧化物的溶液,例如8~12wt%;
[0043]
(2)将载体放置于浸渍液中,浸渍一段时间(例如7~10h),过滤,干燥,焙烧得到负载型催化剂。
[0044]
在本发明中,利用负载型催化剂进行反应,能够避免强氧化剂可能引起的副反应。使用少量的负载型催化剂就能够进行反应,而且其廉价易得,能够降低反应成本。
[0045]
优选地,在步骤2中,2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇与负载型催化剂的摩尔比为(2~10):1,优选为(3~7):1。
[0046]
优选地,在步骤2中,反应温度为30~80℃,优选为50~70℃;和/或,反应压力为
‑
15~5mpa,优选为
‑
5~2mpa。
[0047]
在本发明中,反应温度和反应压力需要保持在一定范围内,使得生成环氧化合物。若反应温度过高或压力过大,原料容易进一步氧化得到酮化合物,若反应温度过低或压力过小,反应不易进行。在本发明中,采用上述范围,使用该负载型催化剂使得原料的转化率较高,生成的副产物较少。
[0048]
步骤3、将步骤1所得的格氏试剂和步骤2所得的环氧丙烷化合物混合,进行反应,得到伏立康唑。
[0049]
在本发明的优选的实施方式中,步骤3具体可包括以下分步骤:
[0050]
步骤3
‑
1、将环氧丙烷化合物溶解于离子液体。
[0051]
优选地,所述离子液体为碱性离子液体。
[0052]
步骤3
‑
2、逐滴加入步骤1所得的格氏试剂,进行反应,得到伏立康唑消旋体。
[0053]
该步骤反应过程为:
[0054][0055]
其中,x表示cl
‑
、br
‑
、i
‑
中的任一者。
[0056]
根据本发明,滴加速度应较缓慢,从而能够使格氏试剂完全溶解在碱性离子液体中。优选地,滴加速度为1~2滴/s。
[0057]
在本发明中,在碱性离子液体中,格氏试剂会形成碳负离子,进攻环氧丙烷化合物中位阻较小环氧上的碳,使其开环形成伏立康唑消旋体。其中伏立康唑(2r,3s)
‑2‑
(2,4
‑
二氟苯基)
‑3‑
(5
‑
氟嘧啶
‑4‑
基)
‑1‑
(1h
‑
1,2,4
‑
三唑
‑1‑
基)
‑2‑
丁醇的量能够控制在70~90%之间。
[0058]
在本发明的优选实施方式中,碱性离子液体选自负载型碱性离子液体或二氧化硅枝载型碱性离子液体。具体地,结构式表示如下:
[0059][0060]
本发明中,所述负载型碱性离子液体优选为聚苯乙烯树脂负载的碱性离子液体。
[0061]
在本发明中,二氧化硅枝载型碱性离子液体可以按以下过程进行制备:
[0062]
(1)将多孔二氧化硅加入到聚丙基三甲氧基硅烷的丙酮溶液中,进行反应;
[0063]
(2)将咪唑加入到步骤1的溶液中,进行反应;
[0064]
(3)将氢氧化钠加入到步骤2的溶液中,进行反应,后处理,得到二氧化硅枝载型碱性离子液体。
[0065]
根据本发明,引入碱性功能基团的离子液体比无机碱或有机碱具有更高的催化活性,且可循环使用。由于其具有较大尺寸的分子结构,其使得不对称加成更容易。
[0066]
在本发明的优选实施方式中,环氧丙烷化合物与碱性离子液体的摩尔比为1:(5~20)。
[0067]
根据本发明,将环氧丙烷化合物与碱性离子液体的摩尔比应在一定范围内,若摩尔比较小,反应速度过快,伏立康唑的占比会减小。若摩尔比较大,反应速度较小,增加反应时间和反应成本。在本发明中,当摩尔比为1:(5~20),优选为1:(10~15)时,伏立康唑的收率在80~90%之间。
[0068]
在本发明中,引入碱性功能基团的离子液体作为碱催化剂,反应转化率高、条件温和,产物易分离,且可循环使用。
[0069]
在本发明的优选实施方式中,格氏试剂和环氧丙烷化合物的摩尔比为1:(1~3)。
[0070]
在本发明中,环氧丙烷化合物的摩尔量稍微大一些,有利于使格氏试剂能够全部加成,而且将格氏试剂的溶液逐步滴加,能够使得反应速度的适宜性,从而使生成的伏立康唑占比较大。
[0071]
本发明中,合成工艺还包括:对伏立康唑消旋体进行拆分,得到伏立康唑,拆分剂优选为光学活性酸,包括但不限于(+)
‑
酒石酸、(+)
‑
樟脑酸、l
‑
(+)
‑
甘氨酸或l
‑
樟脑
‑
10
‑
磺酸及其加成盐。
[0072]
根据本发明第二方面,提供一种伏立康唑或其消旋体,其由以上第一方面的合成工艺或方法制得。
[0073]
为了进一步理解本发明,下面结合实施例对本发明提供的伏立康唑进行描述,本发明的保护范围不受以下实施例的限制。
[0074]
实施例1
[0075]
在装有双口接管的烧瓶中,放置4.8g镁屑。接口的正口处装有滴液漏斗,侧口处装有氯化钙干燥管的回流冷凝管。向瓶中滴加0.2mol 4
‑
(1
‑
溴乙基)
‑5‑
氟嘧啶和302ml甲基四氢呋喃的混合溶液,并水浴加热,温度为50℃,得到格氏试剂的甲基四氢呋喃溶液;
[0076]
称取0.2mol 2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
丙二醇和0.05mol氧化钾
‑
氧化硅负载型催化剂(其中氧化钾的担载量为12wt%),反应温度为65℃,反应压力为2mpa;得到2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
环氧丙烷;
[0077]
取0.1mol以上制得的2
‑
(2,4
‑
二氟苯基)
‑3‑
(1,2,4
‑
三唑
‑1‑
基)
‑
1,2
‑
环氧丙烷溶解于1.2mol二氧化硅枝载型碱性离子液体,逐滴加入100ml所制得的格氏试剂的甲基四氢呋喃溶液,反应结束后,丙酮或乙醇多次洗涤,得到32.8g伏立康唑消旋体,收率93.7%。
[0078]
实施例2
[0079]
取0.05mol实施例1制得的伏立康唑消旋体溶于73.7ml丙酮中,向其中加入13.9g溶于24ml甲醇中的l
‑
(
‑
)
‑
10
‑
樟脑磺酸。所得混合物回流1h,并缓慢冷却至室温,结晶,过滤,干燥,得到伏立康唑的樟脑磺酸盐;
[0080]
将伏立康唑的樟脑磺酸盐加入100ml水和二氯甲烷体积比为1:1的混合物中,并向其中缓慢加入40%氢氧化钠溶液,调节到ph=11
‑
12。分离有机层,干燥,减压除去有机溶剂,异丙醇结晶,干燥,得到13.7g白色的伏立康唑,收率92%,所得伏立康唑的质谱图如图1所示。
[0081]
以上结合具体实施方式和范例性实例对本发明进行了详细说明,不过这些说明并不能理解为对本发明的限制。本领域技术人员理解,在不偏离本发明精神和范围的情况下,可以对本发明技术方案及其实施方式进行多种等价替换、修饰或改进,这些均落入本发明的范围内。
相关技术
网友询问留言
已有1条留言
-
 0158683... 来自[中国] 2023年09月26日 15:07这个网站蛮好的
0158683... 来自[中国] 2023年09月26日 15:07这个网站蛮好的
1