一种氟班色林的合成方法与流程
1.本发明属于有机合成技术领域,具体涉及一种氟班色林的合成方法。
背景技术:
2.氟班色林(flibanserin)由德国勃林格殷格翰制药公司研发,之后转让给美国萌芽制药公司并提交上市申请,于2015年8月18日获得美国食品药品管理局(fda)批准,用于治疗绝经前妇女获得性、广义的机能减退的性欲障碍(hsdd)。氟班色林是一种非激素类药物,对大脑关键性的神经递质起主要作用,能减少抑制性欲的5
‑
羟色胺,以提高刺激性欲的多巴胺水平。氟班色林中文化学名为1
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙基]
‑
2,3
‑
二氢
‑
1h
‑
苯并咪唑
‑2‑
酮,分子式为c
20
h
21
f3n4o,结构式如下:
[0003][0004]
文献报道的氟班色林的合成方法主要有如下几条:
[0005]
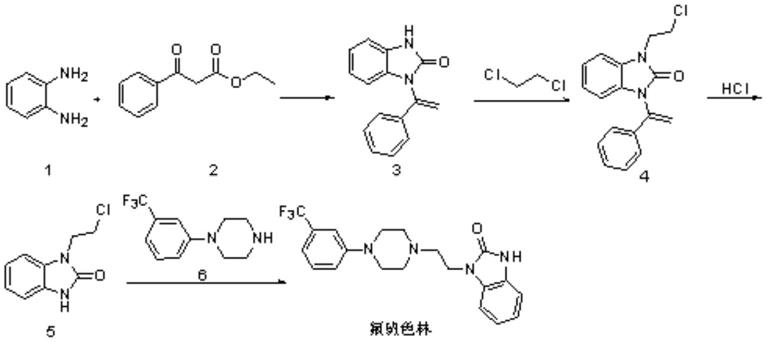
一、原研专利wo199303016报道的合成路线是以邻苯二胺(1)和苯甲酰乙酸乙酯(2)为原料,经关环、重排反应得到咪唑酮中间体(3),经1,2
‑
二氯乙烷的烃基化反应得到氯乙基中间体(4),再用酸脱保护、与哌嗪中间体(6)反应即可制得氟班色林。反应式如下:
[0006][0007]
该路线的第一步合成咪唑酮中间体(3)的反应需要在高温下进行,容易产生热重排副产物;最后一步反应中的氯乙基中间体(5)也容易发生自身的取代反应生成二聚体杂质,导致该路线总体收率不高。
[0008]
二、专利us2003119850公开了一种工业化的合成路线,1
‑
异丙烯基
‑
苯并咪唑
‑2‑
酮(7)与氯代物中间体1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐(8)进行缩合、脱保护一锅法得到氟班色林,反应式如下:
[0009][0010]
中国专利cn111303043报道的合成路线基本与此类似,优化了上述反应式中的两个起始原料的合成方法。
[0011]
三、wo2010128516对原有的合成方法的进行了改进,以邻苯二胺(1)和乙酰乙酸乙酯(10)为原料,关环重排反应得到1
‑
异丙烯基
‑
苯并咪唑
‑2‑
酮(7),接着与二溴乙烷反应得到1,3
‑
二氢
‑1‑
(2
‑
溴乙基)
‑3‑
异丙烯基
‑
2h
‑
苯并咪唑
‑2‑
酮(11),再与二乙醇胺进行胺化反应得到中间体(12),氯代或苯磺酰化反应后与间三氟甲基苯胺环合、脱保护得到氟班色林;此专利还公开了另一种合成方式,1,3
‑
二氢
‑1‑
(2
‑
溴乙基)
‑3‑
异丙烯基
‑
2h
‑
苯并咪唑
‑2‑
酮(11)与哌嗪反应后脱保护、钯催化偶联得到氟班色林。反应式如下:
[0012]
[0013][0014]
该专利报道的合成路线较长,反应控制繁琐,钯催化的重金属偶联反应成本较高,在终产品中容易引起重金属超标等问题。此外中国专利cn108456173报道采用醋酸镍代替醋酸钯进行偶联反应,降低了生产成本,但反应控制严格,也无法解决重金属残留等问题
[0015]
四、中国专利cn104926734公开了另外一种合成方法,以间三氟甲基苯胺(15)、三(2
‑
卤代乙基)胺和邻硝基苯胺为原料,经环化、取代、还原和缩合四步反应得到氟班色林,此路线在最后一步构建苯并咪唑环的反应温度较高,易产生热降解产物,并且在第二步反应中邻硝基苯胺由于邻位的氢键作用,其在进行n烷基化反应时需要较高的温度和额外的催化剂,产品纯化较繁琐。此路线的反应式如下:
[0016][0017]
五、中国专利cn106749038公开的合成方法与上述二中专利us2003119850的路线相似,皆是在最后一步脱保护,不同之处是1
‑
异丙烯基
‑
苯并咪唑
‑2‑
酮(7)先与1,2
‑
二氯乙烷反应,再与哌嗪中间体(6)进行烷基化反应;同时cn107235913、cn106966991、cn107200711公开的路线也与此大体相同,只是烷基化试剂或保护基稍有不同。其合成反应式如下:
[0018][0019]
六、中国专利cn106632066公开了进一步改进的合成方法,以邻苯二胺(1)为起始原料,经环合、n
‑
烷基化反应和脱保护四步反应得到氟班色林,此路线采用原碳酸四乙酯构建苯并咪唑环,减少了原有路线中引入的杂质,但中间体2
‑
乙氧基
‑
1h
‑
苯并咪唑(23)对酸不稳定,后面还有进行两次烷基化反应,增加了反应控制难度,同时原碳酸四乙酯相比其他的羰基环合试剂,成本较高。其反应式如下:
[0020][0021]
七、中国专利cn109232434公开的合成方法与上述六的路线相类似,不同之处是中间体2
‑
乙氧基
‑
1h
‑
苯并咪唑(23)直接与氯代物中间体(8)进行缩合反应,再脱保护得到氟班色林,此路线设计更加合理。反应式如下:
[0022][0023]
八、期刊(化学通报,2018,81(11),1048
‑
1051)报道了另外一种合成路线,利用醋酸碘苯促进的n
‑
苯基羟胺(26)、苯甲醛(27)和三甲基氰硅烷的一锅法反应来构建苯并咪唑酮结构(28),接着进行烷基化、脱保护、胺化三步反应得到氟班色林,此路线所用原料和试剂价格较高,收率适中,总体成本较高。反应式如下:
[0024][0025]
综述所述,目前报道的氟班色林合成路线存在起始原料难获得、反应步骤多、副反应难控制及成本较高等弊端,需要不断优化或改进合成方法,寻找一条操作简便、经济环保的工艺路线。
技术实现要素:
[0026]
本发明的目的在于提供一种合成步骤少、经济环保、适合工业化生产的氟班色林的合成方法。
[0027]
本发明解决其技术问题所采用的技术方案是:一种氟班色林的合成方法,包括以下步骤:
[0028]
1)将氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂在有机溶剂中加热反应,完成后经后处理得到2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸甲酯粗品,然后在甲醇中用10wt%氢氧化钠水溶液室温碱解,酸化得到2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸;
[0029]
2)将2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱在有机溶剂中加热反应,经后处理得到的粗品用乙醇重结晶,制得氟班色林。
[0030]
进一步地,所述步骤1)中氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂的摩尔比为1.0
‑
1.2:1.0:1.9
‑
2.1:0.005
‑
0.02,优选为1.1:1.0:2.0:0.01。
[0031]
进一步地,所述步骤1)中碱为氢氧化钠、氢氧化钾、碳酸钠、碳酸钾、碳酸钙、三乙胺、二异丙基乙胺、吡啶、n
‑
甲基吗啉、三乙烯二胺、二氮杂二环中的一种,优选为碳酸钾或二异丙基乙胺。
[0032]
进一步地,所述步骤1)中有机溶剂为甲苯、氯苯、二甲苯、二氧六环、n,n
‑
二甲基甲酰胺、二甲亚砜中的一种,优选为二氧六环或n,n
‑
二甲基甲酰胺。
[0033]
进一步地,所述步骤1)中催化剂为碘化钠、碘化钾或四丁基碘化胺,优选为碘化钾。
[0034]
进一步地,所述步骤2)中2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱的摩尔比为1.0:1.0
‑
1.2:1.5
‑
3.0,优选为1.0:1.05:2.0。
[0035]
进一步地,所述步骤2)中有机溶剂为甲苯、二甲苯、二氧六环、n,n
‑
二甲基甲酰胺、二甲亚砜中的一种,优选为甲苯或二氧六环。
[0036]
进一步地,所述步骤2)中有机碱为三乙胺、二氮杂二环、n
‑
甲基吗啉、二异丙基乙
胺中的一种。
[0037]
本发明的合成路线如下:
[0038][0039]
本发明具有以下有益效果:本发明以氨茴酸甲酯为起始原料,先与1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐在碱的作用下进行胺基的烷基化反应,接着进行碱解脱甲酯,最后在叠氮磷酸二苯酯(dppa)的作用下发生schmidt反应得到活泼的异氰酸酯中间体,继而发生分子内环合得到氟班色林。该方法与现有技术相比,合成步骤短,反应位点少,降低了副反应的发生,中间体和目标物的收率均较高;本方法的操作简便,后处理工艺简洁,对反应设备要求低,原料价廉易得,经济环保,适合工业化生产。
具体实施方式
[0040]
以下是本发明的具体实施例,对本发明的技术方案做进一步描述,但是本发明的保护范围并不限于这些实施例。凡是不背离本发明构思的改变或等同替代均包括在本发明的保护范围之内。
[0041]
实施例1
[0042]
氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碳酸钾和催化剂碘化钾在二氧六环中回流48小时,氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂的摩尔比为1.1:1.0:2.0:0.01,反应完成后过滤,滤液减压浓缩,残留物溶解到1.0l甲醇中,加入10wt%氢氧化钠水溶液,室温搅拌4小时,减压浓缩除掉甲醇,残留物用浓盐酸调ph至5
‑
6,在5
‑
10℃搅拌2小时,抽滤,依次用冷水和异丙醇洗涤,干燥,得到类白色固体产品2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸,收率84.9%。
[0043]2‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸溶解到有机溶剂二氧六环中,依次加入有机碱三乙胺和叠氮磷酸二苯酯,2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱的摩尔比为1.0:1.05:2.0,反应混合物室温下搅拌3小时,然后加热至回流反应6小时,减压浓缩除掉反应溶剂,加入5wt%氢氧化钠水溶液和乙酸乙酯,分液,有机相用无水硫酸钠干燥浓缩,所得残留物用乙醇重结晶得到目标物氟班色林,收率72.4%。
[0044]
实施例2
[0045]
氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、二异丙基乙胺和催化剂碘化钠在n,n
‑
二甲基甲酰胺中回流48小时,氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂的摩尔比为1.2:1.0:1.9:0.007,反应完成后过
滤,滤液减压浓缩,残留物溶解到1.0l甲醇中,加入10wt%氢氧化钠水溶液,室温搅拌4小时,减压浓缩除掉甲醇,残留物用浓盐酸调ph至5
‑
6,在5
‑
10℃搅拌2小时,抽滤,依次用冷水和异丙醇洗涤,干燥,得到类白色固体产品2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸,收率81.5%。
[0046]2‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸溶解到有机溶剂甲苯中,依次加入有机碱二异丙基乙胺和叠氮磷酸二苯酯,2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱的摩尔比为1.0:1.1:2.5,反应混合物室温下搅拌3小时,然后加热至回流反应6小时,减压浓缩除掉反应溶剂,加入5wt%氢氧化钠水溶液和乙酸乙酯,分液,有机相用无水硫酸钠干燥浓缩,所得残留物用乙醇重结晶得到目标物氟班色林,收率71.7%。
[0047]
实施例3
[0048]
氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、三乙胺和催化剂四丁基碘化胺在n,n
‑
二甲基甲酰胺中回流48小时,氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂的摩尔比为1.0:1.0:2.1:0.02,反应完成后过滤,滤液减压浓缩,残留物溶解到1.0l甲醇中,加入10wt%氢氧化钠水溶液,室温搅拌4小时,减压浓缩除掉甲醇,残留物用浓盐酸调ph至5
‑
6,在5
‑
10℃搅拌2小时,抽滤,依次用冷水和异丙醇洗涤,干燥,得到类白色固体产品2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸,收率75.9%。
[0049]2‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸溶解到有机溶剂二甲亚砜中,依次加入有机碱二氮杂二环和叠氮磷酸二苯酯,2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱的摩尔比为1.0:1.2:1.5,反应混合物室温下搅拌3小时,然后加热至回流反应6小时,减压浓缩除掉反应溶剂,加入5wt%氢氧化钠水溶液和乙酸乙酯,分液,有机相用无水硫酸钠干燥浓缩,所得残留物用乙醇重结晶得到目标物氟班色林,收率69.4%。
[0050]
实施例4
[0051]
氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碳酸钠和催化剂碘化钾在n,n
‑
二甲基甲酰胺中回流48小时,氨茴酸甲酯、1
‑
(2
‑
氯乙基)
‑4‑
[3
‑
(三氟甲基)苯基]哌嗪二盐酸盐、碱和催化剂的摩尔比为1.15:1.0:2.0:0.005,反应完成后过滤,滤液减压浓缩,残留物溶解到1.0l甲醇中,加入10wt%氢氧化钠水溶液,室温搅拌4小时,减压浓缩除掉甲醇,残留物用浓盐酸调ph至5
‑
6,在5
‑
10℃搅拌2小时,抽滤,依次用冷水和异丙醇洗涤,干燥,得到类白色固体产品2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸,收率79.9%。
[0052]2‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸溶解到有机溶剂n,n
‑
二甲基甲酰胺中,依次加入有机碱n
‑
甲基吗啉和叠氮磷酸二苯酯,2
‑
[2
‑
[4
‑
[3
‑
(三氟甲基)苯基]哌嗪
‑1‑
]乙胺基]苯甲酸、叠氮磷酸二苯酯和有机碱的摩尔比为1.0:1.0:3.0,反应混合物室温下搅拌3小时,然后加热至回流反应6小时,减压浓缩除掉反应溶剂,加入5wt%氢氧化钠水溶液和乙酸乙酯,分液,有机相用无水硫酸钠干燥浓缩,所得残留物用乙醇重结晶得到目标物氟班色林,收率70.5%。
[0053]
本发明不局限于上述实施方式,任何人应得知在本发明的启示下作出的结构变
化,凡是与本发明具有相同或相近的技术方案,均落入本发明的保护范围之内。
[0054]
本发明未详细描述的技术、形状、构造部分均为公知技术。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1