一种高纯度双-乙基己氧苯酚甲氧苯基三嗪的制备方法与流程
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法
技术领域
1.本发明属于化工合成技术领域,具体涉及一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法。
背景技术:
2.双
‑
乙基己氧苯酚甲氧苯基三嗪,其化学名称为2,4
‑
双[[4
‑
(2
‑
乙基
‑
己氧基)
‑2‑
羟基)]
‑
苯基]
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪,商品名为bemotrizinol,英文名称为bis
‑
ethylhexyloxyphenol methoxyphenyl triazine(tinosorb s),双
‑
乙基己氧苯酚甲氧苯基三嗪是一种黄色粉末状物质,cas号为187393
‑
00
‑
6,分子式为c
38
h
49
n3o5,相对分子质量为627.8。双
‑
乙基己氧苯酚甲氧苯基三嗪是一种新型的高效广谱油溶性紫外线吸收剂,是2000年瑞士ciba specialty chemical公司研发出来,能全面防护uva和uvb,吸收波长在290
‑
370nm之间;其分子量大,不易被皮肤吸收,用量小但防护能力强且光稳定性极强,还可以作为其他化学防晒剂的稳定剂,并且与其他防晒剂的相溶性较强。
[0003]
目前国内外报道的双
‑
乙基己氧苯酚甲氧苯基三嗪的合成方法中,基本都是通过2
‑
(4
‑
甲氧基苯基)
‑
4,6
‑
二氯
‑
1,3,5
‑
三嗪和间苯二酚在路易斯酸催化下发生付克酰基化反应得到中间体2,4
‑
双(2,4
‑
二羟基苯基)
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
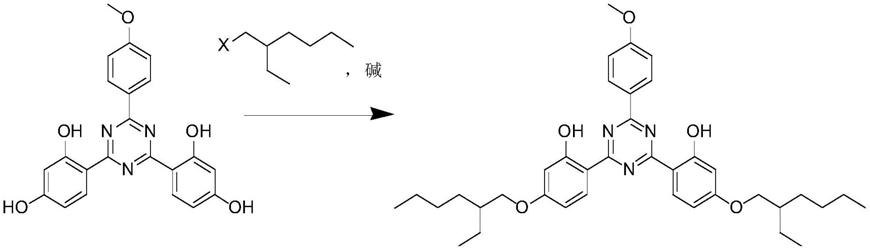
三嗪,再在碱的催化下,与卤代烷烃进行烷基化反应而得到,反应方程式如下:
[0004][0005]
如中国专利文献cn201680027923.3中公开了一种用于醚化双
‑
间苯二酚基三嗪的新方法,该技术方案中是用2,4
‑
双(2,4
‑
二羟基苯基)
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和3
‑
氯甲基庚烷在dmf溶剂中,在碱催化下,可以获得产率为78
‑
87%,纯度为97.1
‑
96.8%的双
‑
乙基己氧苯酚甲氧苯基三嗪。再如美国专利文献us5955060中公开的方法,该技术方案中是用2,4
‑
双(2,4
‑
二羟基苯基)
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和3
‑
溴甲基庚烷在乙二醇甲醚溶剂中,在液碱催化下反应获得产率为78.4%的双
‑
乙基己氧苯酚甲氧苯基三嗪。
[0006]
上述现有技术的制备方法中,在烷基化反应过程中会不可避免的出现烷基化反应不充分,或者过度反应的现象,从而使产物中出现单烷基化、多烷基化副产物,通常反应结束时,目标产物转化率为90%左右,单烷基化产物剩余0.5
‑
2%,三烷基化产物有5
‑
8%,很难获得高纯度产物。而且由于单烷基化、多烷基化副产物的存在,使得后续精制提纯过程中不得不增加溶剂用量或增加重结晶次数,从而使得产率也偏低。
技术实现要素:
[0007]
针对上述现有技术中的不足,本发明的目的是提供一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,解决现有技术烷基化反应过程中会不可避免的出现烷基化反应不充分,或者过度反应的现象,从而使产物中出现单烷基化、多烷基化副产物等问题。
[0008]
为解决现有技术中的上述问题,本发明是通过如下技术方案实现的:
[0009]
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,包括如下步骤:
[0010]
s1,向反应容器中加入2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和3
‑
乙基己氧基苯酚混合,之后加入有机溶剂、催化剂进行充分反应,得到反应液;
[0011]
s2,加水猝灭步骤s1中所述的反应液,油相经水洗、脱溶、重结晶、过滤、烘干后制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪。
[0012]
进一步地,步骤s1中,所述3
‑
乙基己氧基苯酚的具体合成方法如下:
[0013]
s11,向反应容器中加入摩尔配比为1:1.2的间苯二酚和碳酸钾,然后加入dmf,所述dmf的用量为所述间苯二酚质量的3.5~4.5倍,连续搅拌并升温至90~110℃;
[0014]
s12,在3~4h内,向步骤s11中滴加溴代异辛烷,所述溴代异辛烷用量与所述间苯二酚用量的摩尔配比为1.2:1,滴加完毕后继续保温反应3~5h;然后降温至40℃以下,经过滤,滤液减压蒸馏出dmf等步骤后,向残留液中加入甲苯,所述甲苯的用量为所述间苯二酚质量的3.5~4.5倍,之后水洗,并调节ph至中性;最后用水洗至水层颜色为无色透明为止;
[0015]
s13,步骤s12中,剩余油层先蒸馏并回收甲苯,然后继续升温减压蒸馏,在真空度50~100pa的环境下,接收140~150℃的馏分,得到浅黄色透明液体,即为3
‑
乙基己氧基苯酚。
[0016]
进一步地,步骤s1中,所述2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪、所述3
‑
乙基己氧基苯酚和所述催化剂用量的摩尔配比为1:(2.0~2.6):(2.0~2.6)。
[0017]
更进一步地,步骤s1中,所述2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪、所述3
‑
乙基己氧基苯酚和所述催化剂用量的摩尔配比为1:(2.1~2.4):(2.2~2.4)。
[0018]
进一步地,步骤s1中,所述有机溶剂的用量为所述2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪质量的8~10倍。
[0019]
进一步地,步骤s1中,加入有机溶剂后,升温至40~50℃,然后分次加入催化剂;待催化剂加完之后于50
‑
60℃保温反应1.5~2h,最后升温至60~70℃进行保温反应2~3h,得到反应液。
[0020]
进一步地,步骤s1中,所述有机溶剂为二氯甲烷、1,2
‑
二氯乙烷、三氯甲烷、氯苯、硝基甲烷、硝基甲苯和二硫化碳中的一种或多种组合。
[0021]
进一步地,步骤s1中,所述催化剂为路易斯酸。
[0022]
更进一步地,所述路易斯酸为三氯化铝、三氯化铁,氯化锌和三氟化硼中的一种或多种组合。
[0023]
进一步地,步骤s2中,加水猝灭步骤s1中所述的反应液具体方法如下:待步骤s1中所述的反应液降温至40℃以下时,向反应容器中缓慢加入水,并且控制温度在60℃以下;之后保持温度在60℃搅拌30~45min,静置,去掉水层,得到油相。
[0024]
本发明中选用原料2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和3
‑
乙基己氧基苯酚,在催化剂的作用下生成目标产物双
‑
乙基己氧苯酚甲氧苯基三嗪。具体的反应机理如
下:
[0025][0026]
由于3
‑
乙基己氧基苯酚中烷基的数量是固定的,因此,可以大幅度减少单烷基化产物(式(ⅰ)所示)、三烷基化产物(式(ⅱ)所示)的产生,获得纯度更高的目标产物,本发明中,催化剂是分批次加入反应体系中的,这样做的好处在于控制反应放热,使原料反应完全;加入有机溶剂后,进行梯度升温,可以使反应更加温和,达到减少副反应的目的。
[0027][0028]
与现有技术相比,本发明具有如下优点:
[0029]
1)本发明中选用原料2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和3
‑
乙基己氧基苯酚,在催化剂的作用下直接一步生成目标产物双
‑
乙基己氧苯酚甲氧苯基三嗪,相比于传统的两步法合成工艺,本发明反应简单,高效,所制得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率在83%以上,产品纯度达到99%以上;整个操作过程更加方便、安全,增加了相应经济效益,并减少了环保压力;
[0030]
2)由于本发明中反应原料3
‑
乙基己氧基苯酚中烷基的数量是固定的,因此,可以大幅度减少单烷基化产物、三烷基化产物的产生,获得纯度更高的目标产物,本发明中,催化剂是分批次加入反应体系中的,这样做的好处在于控制反应放热,使原料反应完全;加入有机溶剂后,进行梯度升温,可以使反应更加温和,达到减少副反应的目的。
附图说明
[0031]
为了更清楚地说明本发明实施例中的技术方案,下面将对实施例描述中所需要使用的附图作简单的介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
[0032]
图1为本发明制备工艺的反应原理图;
[0033]
图2为实施例1中制得的双
‑
乙基己氧苯酚甲氧苯基三嗪液相色谱图。
具体实施方式
[0034]
下面将结合具体实施例对本发明的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0035]
本发明所使用的原料和设备,如无特别说明,均为市售获得。
[0036]
本发明首先合成了3
‑
乙基己氧基苯酚,具体合成方法如下:
[0037]
s11,向反应容器中加入55g间苯二酚、83g碳酸钾和200g dmf,连续搅拌并升温至90~110℃;
[0038]
s12,在3~4h内,向步骤s11中滴加120g溴代异辛烷,滴加完毕后继续保温反应3~5h;然后降温至40℃以下,经过滤,滤液减压蒸馏出dmf等步骤后,向残留液中加入200g甲苯,之后水洗,并调节ph至中性;最后用水洗至水层颜色为无色透明为止;
[0039]
s13,步骤s12中,剩余油层先蒸馏并回收甲苯,然后继续升温减压蒸馏,在真空度50~100pa的环境下,接收140~150℃的馏分,得到85.6g浅黄色透明液体,即为3
‑
乙基己氧基苯酚,收率77%,纯度≥98.0%。
[0040]
上述方法合成的3
‑
乙基己氧基苯酚作为反应原料,用于高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备。
[0041]
实施例1
[0042]
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,包括如下步骤:
[0043]
s1,向1l反应容器中加入51.2g 2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和102.12g 3
‑
乙基己氧基苯酚混合,之后加入450g氯苯、升温至40~50℃,然后分4次加入61.18g无水三氯化铝;待无水三氯化铝加完之后于50
‑
60℃保温反应1.5~2h,最后升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液;
[0044]
s2,待步骤s1中所述的反应液降温至40℃以下时,向反应器中缓慢加入200~300g水,加水过程控制温度低于60℃,之后保持温度在60℃搅拌30~40min,静置,去掉水层,油层再分别用200~300g水洗2次;油相经水洗、脱溶、重结晶、过滤、烘干后制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品115.3g,收率91.9%,所得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品经液相色谱检测,纯度为99.7%,液相色谱图见图2,其分析结果如下表1所示:
[0045]
表1高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品液相色谱检测成分表
[0046][0047]
双
‑
乙基己氧苯酚甲氧苯基三嗪成品采用高效液相色谱法检测,具体方法如下:准确制得的称双
‑
乙基己氧苯酚甲氧苯基三嗪样品0.05g于50ml容量瓶中,用40ml 1,4
‑
二恶烷超声溶解,冷却至室温后用1,4
‑
二恶烷定容,混匀,取5ml于50ml容量瓶中,用稀释溶剂(1,4
‑
二恶烷:水=80:20)定容,混匀后,过孔径0.22μm滤膜,取10μl溶液进行上样分析;高效液相色谱购自美国安捷伦公司,仪器型号为agilent1220,装配有紫外检测器,具体液相
色谱条件如下:
[0048]
色谱柱:usp l14
‑
c18(4.6mm
×
150mm,5μm);流动相:流动相a为0.06%甲酸铵水溶液,用甲酸调节ph至4.6,流动相b为1,4
‑
二恶烷,进行梯度洗脱,梯度程序洗脱见表2;流速0.8ml/min,柱温35℃,进样量10μl,检测波长332nm。
[0049]
表2梯度程序洗脱表
[0050][0051][0052]
实施例2
[0053]
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,包括如下步骤:
[0054]
s1,向1l反应容器中加入51.2g 2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和106.56g 3
‑
乙基己氧基苯酚混合,之后加入500g 1,2
‑
二氯乙烷、升温至40~50℃,然后分4次加入77.76g无水三氯化铁;待无水三氯化铁加完之后于50
‑
60℃保温反应1.5~2h,最后升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液;
[0055]
s2,待步骤s1中所述的反应液降温至40℃以下时,向反应器中缓慢加入200~300g水,加水过程控制温度低于60℃,之后保持温度在60℃搅拌30~40min,静置,去掉水层,油层再分别用200~300g水洗2次;油相经水洗、脱溶、重结晶、过滤、烘干后制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品109.5g,收率87.3%,所得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品经液相色谱检测,纯度为99.4%。
[0056]
实施例3
[0057]
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,包括如下步骤:
[0058]
s1,向1l反应容器中加入51.2g 2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和93.24g 3
‑
乙基己氧基苯酚混合,之后加入300g硝基甲苯,升温至40~50℃,然后将29.92g三氟化硼溶解于110g二硫化碳溶液中,1~1.5h内滴加到上述反应容器中;待滴加完之后于50
‑
60℃保温反应1.5~2h,最后升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液;
[0059]
s2,待步骤s1中所述的反应液降温至40℃以下时,向反应器中缓慢加入200~300g水,加水过程控制温度低于60℃,之后保持温度在60℃搅拌30~40min,静置,去掉水层,油层再分别用200~300g水洗2次;油相经水洗、脱溶、重结晶、过滤、烘干后制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品106.6g,收率85.0%,所得的高纯度双
‑
乙基己氧苯酚
甲氧苯基三嗪成品经液相色谱检测,纯度为99.5%。
[0060]
实施例4
[0061]
一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法,包括如下步骤:
[0062]
s1,向1l反应容器中加入51.2g 2,4
‑
二氯
‑6‑
(4
‑
甲氧基苯基)
‑
1,3,5
‑
三嗪和115.44g 3
‑
乙基己氧基苯酚混合,之后加入500g 1,2
‑
二氯乙烷、二氯甲烷和氯苯的混合液,升温至40~50℃,然后分4次加入31.92g无水三氯化铝和32.64g氯化锌的混合物;待上述混合物加完之后于50
‑
60℃保温反应1.5~2h,最后升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液;
[0063]
s2,待步骤s1中所述的反应液降温至40℃以下时,向反应器中缓慢加入200~300g水,加水过程控制温度低于60℃,之后保持温度在60℃搅拌30~40min,静置,去掉水层,油层再分别用200~300g水洗2次;油相经水洗、脱溶、重结晶、过滤、烘干后制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品104.83g,收率83.6%,所得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品经液相色谱检测,纯度为99.1%。
[0064]
对比例1
[0065]
高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法与实施例1基本相似,不同之处在于,无水三氯化铝一次性加入。
[0066]
反应结束后,制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品103.2g,收率82.3%,所得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品经液相色谱检测,纯度为99.1%。
[0067]
对比例2
[0068]
高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制备方法与实施例1基本相似,不同之处在于,不进行梯度升温反应,待原料加完之后,直接升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液。
[0069]
反应结束后,制得所述高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品100.95g,收率80.5%,所得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品经液相色谱检测,纯度为88.5%。
[0070]
由实施例1~4的结果可以看出,采用本发明新的合成工艺,使制得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率在83%以上,产品纯度达到99%以上,所制得的产品收率和纯度均高于背景技术中描述的传统方法合成工艺;
[0071]
对比例1与实施例1相比,区别在于,催化剂无水三氯化铝一次性加入。所制得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率82.3%,纯度为99.1%,实施例1中的收率较高,这是因为实施例1中催化剂是分批次加入反应体系中的,这样做的好处在于控制反应放热,使原料反应完全,从而提高双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率;
[0072]
对比例2与实施例1相比,区别在于,不进行梯度升温反应,待原料加完之后,直接升温至60~70℃进行保温反应2~3h至没有氯化氢气体产生为止,得到反应液。所制得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率80.5%,纯度为88.5%,由于对比例2没有进行梯度升温,导致反应不完全,发生较多的副反应,因此,制得的高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪成品收率和纯度都偏低。
[0073]
综上所述,本发明的目的是提供一种高纯度双
‑
乙基己氧苯酚甲氧苯基三嗪的制
备方法,解决现有技术烷基化反应过程中会不可避免的出现烷基化反应不充分,或者过度反应的现象,从而使产物中出现单烷基化、多烷基化副产物等问题。由于3
‑
乙基己氧基苯酚中烷基的数量是固定的,因此,可以大幅度减少单烷基化产物、三烷基化产物的产生,获得纯度更高的目标产物。
[0074]
以上实施例,仅为本发明的具体实施方式,用以说明本发明的技术方案,而非对其限制,本发明的保护范围并不局限于此,尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,其依然可以对前述实施例所记载的技术方案进行修改或可轻易想到变化,或者对其中部分技术特征进行等同替换;而这些修改、变化或者替换,并不使相应技术方案的本质脱离本发明实施例技术方案的精神和范围,都应涵盖在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1