一种脑保护剂的制备工艺的制作方法
1.本发明属于医药技术领域,具体涉及一种脑保护剂的制备工艺。
背景技术:
2.依达拉奉,化学名为3
‑
甲基
‑1‑
苯基
‑2‑
吡唑啉
‑5‑
酮,由日本三菱田边制药公司研发,于2001年6月在日本上市,目前国内已有多家仿制药上市。本品用于治疗脑梗死急性期相关神经症状;2015年6月日本批准新增适应证肌萎缩侧索硬化症(als);2017年5月美国批准用于治疗als。
3.依达拉奉,化学名为3
‑
甲基
‑1‑
苯基
‑2‑
吡唑啉
‑5‑
酮,由日本三菱田边制药公司研发,于2001年6月在日本上市,目前国内已有多家仿制药上市。本品用于治疗脑梗死急性期相关神经症状;2015年6月日本批准新增适应证肌萎缩侧索硬化症(als);2017年5月美国批准用于治疗als。
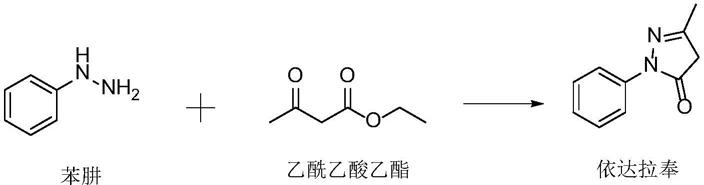
4.依达拉奉的制备工艺主要是苯肼和丁酮酰胺或乙酰乙酸乙酯反应,乙酰乙酸乙酯更容易与苯肼反应且更易得到,价格也更便宜,因此现有技术主要采用苯肼与乙酰乙酸乙酯的制备工艺。苯肼与乙酰乙酸乙酯反应路线如下:
[0005][0006]
该路线虽然可以一锅完成,但反应过程中会先生成过渡态杂质,再生成依达拉奉,实际反应中为了避免由过渡态生成依达拉奉的过程中依然有过渡态杂质生成,通常需要在生成过渡态后进行淬灭反应,由于淬灭反应需要一段时间,因此很难精确控制淬灭反应的时机,难免因淬灭提前或不及时导致反应不充分或杂质残留,从而影响最终产品的收率和纯度。
[0007][0008]
比如文献【殷晓华.依达拉奉的合成工艺改进[j].化学工程与装备,2013,】使用苯肼与乙酰乙酸乙酯在无水乙醇中回流反应7小时,降温析晶,得到依达拉奉收率为50.6%。
[0009]
文献【姚爱平.依达拉奉的合成[j].中国现代应用药学杂志,2003,20(7)】使用苯肼与乙酰乙酸乙酯在乙酸回流反应中,缩短反应时间至4小时,收率为50.3%。
[0010]
专利cn109608398a公开了一种依达拉奉的制备方法,苯肼与乙酰乙酸乙酯先在无水乙醇中室温反应,然后在乙酸中回流反应,总体反应时间较长,且粗品经两次乙醇精制和两次乙酸乙酯精制,两次乙醇精制收率及两次乙酸乙酯精制收率均在50%左右。
[0011]
综上所述,由于过渡态杂质的存在,苯肼与乙酰乙酸乙酯的一锅反应并不具备大多数一锅反应收率高的优点。
技术实现要素:
[0012]
为了克服现有技术中的不足,本发明提供了一种采用新的原料制备收率和纯度较高的脑保护剂依达拉奉的工艺。
[0013]
一种脑保护剂的制备工艺,包括以下步骤:
[0014]
(1)化合物i、苯胺及乙醇钠在室温下接触生成化合物ii;
[0015]
(2)化合物ii在氢氧化钠及对甲苯磺酰氯的作用下生成依达拉奉;
[0016]
反应路线如下:
[0017][0018]
化合物i为(3z)
‑3‑
羟基亚胺丁酸乙酯,来源于河南天孚化工有限公司,纯度99%,申请人在实验中发现,该化合物可与苯胺在室温下反应生成依达拉奉的前体化合物ii,进而生成依达拉奉,从而解决了现有技术中采用乙酰乙酸乙酯及苯肼为起始原料一步法合成依达拉奉面临的过渡态杂质的问题。
[0019]
一种脑保护剂的制备工艺,具体的,包括以下步骤:
[0020]
(1)乙醇溶剂中加入苯胺和化合物i,加入乙醇钠,控制室温反应,反应结束后加入盐酸调节ph至4.5
‑
6.0,加入纯化水析晶,得到化合物ii;
[0021]
(2)将化合物ii溶于二氯甲烷溶剂,加入氢氧化钠及对甲苯磺酰氯,升温至回流搅拌,反应1小时后结束,完毕加入水,二氯甲烷萃取,减压蒸除溶剂,加入乙醇,降温析晶,得依达拉奉。
[0022]
其中,步骤(1)中所述苯胺与化合物i的摩尔比为1.1:1,所述乙醇与化合物i的体积质量比为4
‑
10:1;
[0023]
步骤(1)中所述乙醇钠与化合物i的摩尔比为1.1
‑
1.5:1;
[0024]
步骤(1)中所述纯化水与化合物i的体积质量比为6
‑
12:1;
[0025]
步骤(2)中所述二氯甲烷与化合物ii的体积质量比为6
‑
12:1;
[0026]
步骤(2)中所述氢氧化钠与化合物ii的摩尔比为1.5
‑
2.5:1;
[0027]
步骤(2)中所述对甲苯磺酰氯与化合物ii的摩尔比为1.1
‑
1.5:1;
[0028]
步骤(2)中所述乙醇与化合物ii的体积质量比为4
‑
12:1。
[0029]
本发明取得以下有益效果:
[0030]
采用新的反应原料,先生成中间体化合物ii,再生成依达拉奉,反应分两步进行,解决了乙酰乙酸乙酯和苯肼为原料先生成过渡态再生成依达拉奉可控性差的问题,提高了
反应收率和纯度。
具体实施方式
[0031]
实施例1依达拉奉的制备
[0032]
将14.52g化合物i和10.24g苯胺加入到58ml乙醇溶剂中,加入7.49g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至4.5,加入纯化水87ml,室温下搅拌析晶2小时,得化合物ii 17.74g,收率92.3%。
[0033]
将19.22g化合物ii加入到115ml二氯甲烷溶剂中,加入6.0g氢氧化钠及20.97g对甲苯磺酰氯,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入77ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉15.96g,收率91.6%,纯度99.92%。
[0034]
实施例2依达拉奉的制备
[0035]
将14.52g化合物i和10.24g苯胺加入到87ml乙醇溶剂中,加入8.17g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至5.0,加入纯化水116ml,室温下搅拌析晶2小时,得化合物ii 17.45g,收率90.8%。
[0036]
将19.22g化合物ii加入到154ml二氯甲烷溶剂中,加入8.0g氢氧化钠及22.88g对甲苯磺酰氯,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入135ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉15.92g,收率91.4%,纯度99.89%。
[0037]
实施例3依达拉奉的制备
[0038]
将14.52g化合物i和10.24g苯胺加入到116ml乙醇溶剂中,加入8.85g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至5.5,加入纯化水145ml,室温下搅拌析晶2小时,得化合物ii 17.68g,收率92.0%。
[0039]
将19.22g化合物ii加入到192ml二氯甲烷溶剂中,加入10.0g氢氧化钠及24.78g对甲苯磺酰氯,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入192ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉15.66g,收率89.9%,纯度99.90%。
[0040]
实施例4依达拉奉的制备
[0041]
将14.52g化合物i和10.24g苯胺加入到145ml乙醇溶剂中,加入10.21g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至6.0,加入纯化水174ml,室温下搅拌析晶2小时,得化合物ii 17.78g,收率92.5%。
[0042]
将19.22g化合物ii加入到230ml二氯甲烷溶剂中,加入8.0g氢氧化钠及28.60g对甲苯磺酰氯,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入230ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉15.99g,收率91.8%,纯度99.90%。
[0043]
对比例1
[0044]
将14.52g化合物i和10.24g苯胺加入到116ml乙醇溶剂中,加入8.85g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至4.0,加入纯化水145ml,室温下搅拌析晶2小时,得化合物ii 14.79g,收率84.9%。
[0045]
对比例2
[0046]
将14.52g化合物i和10.24g苯胺加入到116ml乙醇溶剂中,加入8.85g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至6.5,加入纯化水145ml,室温下搅拌析晶2小时,得化合物ii 15.03g,收率86.3%。
[0047]
对比例3
[0048]
将14.52g化合物i和10.24g苯胺加入到116ml乙醇溶剂中,加入8.85g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至5.5,加入纯化水145ml,室温下搅拌析晶2小时,得化合物ii 17.68g,收率92.0%。
[0049]
将19.22g化合物ii加入到192ml二氯甲烷溶剂中,加入24.78g对甲苯磺酰氯,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入192ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉10.85g,收率62.3%,纯度96.8%。
[0050]
对比例4
[0051]
将14.52g化合物i和10.24g苯胺加入到116ml乙醇溶剂中,加入8.85g乙醇钠,控制室温反应,hplc监测反应结束后加入浓盐酸调节ph至5.5,加入纯化水145ml,室温下搅拌析晶2小时,得化合物ii 17.68g,收率92.0%。
[0052]
将19.22g化合物ii加入到192ml二氯甲烷溶剂中,加入10.0g氢氧化钠,回流搅拌1小时,加入150ml纯化水,加入150ml二氯甲烷萃取,有机相减压蒸除溶剂,加入192ml乙醇,降温至0
‑
5℃析晶2小时,得依达拉奉8.64g,收率49.6%,纯度95.4%。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1