二氟苯环丙胺类化合物及其制备方法和应用
1.本发明属于药物化学领域,具体涉及二氟苯环丙胺类化合物及其制备方法,并将其应用于以组蛋白赖氨酸特异性去甲基化酶1(lsd1)为靶点的抗肿瘤药物的开发中。
背景技术:
2.赖氨酸特异性去甲基化酶1(lsd1)是第一个被发现的组蛋白去甲基化酶,通过黄素腺嘌呤二核苷酸(fad)依赖型机制特异性脱去单甲基化和二甲基化h3k4和h3k9位点上的甲基基团,从而调节基因表达和转录活性。目前研究证实,lsd1在多种肿瘤细胞中过表达。lsd1可通过组蛋白的去甲基化作用激活或抑制的染色质结构域从而调控基因的表达,并且通过影响细胞增殖、分化中所必需因子的表达,从而调控肿瘤的发生与发展。因此,lsd1作为一个有吸引力的抗癌药物靶点,得到越来越多药物化学家的关注。
3.由于lsd1属于胺氧化酶家族,与单胺氧化酶(maos)a和b在结构和序列上具有相似性,所以在早期研究中,maos抑制剂被用来筛选具有lsd1抑制活性的骨架结构,其中苯环丙胺(tcp)活性最好。目前已经报道的lsd1抑制剂中,有6种具有tcp结构的lsd1抑制剂:tcp、ory
‑
1001,gsk
‑
2879552,incb059872,img
‑
7289和ory
‑
2001,已用于临床试验研究。因此,tcp作为一种lsd1抑制剂的药效团得到了越来越多人的关注,基于tcp的结构拓展和多样性小分子化合物的合成,对于开发新型的lsd1小分子抑制剂具有一定的指导意义。
技术实现要素:
4.有鉴于此,为开发利用现有的临床药物资源,本发明有必要提供一种新型的二氟苯环丙胺类化合物及其制备方法和应用。
5.因此,本发明的一个目的在于提供一类二氟苯环丙胺类化合物及其制备方法,从而为寻找一类新的基于lsd1靶点的抗肿瘤药物开辟一条新途径。
6.本发明的另一目的在于提供一种3,4
‑
二氟苯环丙胺类衍生物或其药学可接受的盐,用于制备lsd1抑制剂。
7.为实现上述目的,本发明提供一种二氟苯环丙胺类化合物,其为3,4
‑
二氟苯环丙胺类衍生物或其药学可接受的盐,其中,所述3,4
‑
二氟苯环丙胺类衍生物的通式如下所示:
[0008][0009]
其中,所述通式中的基团r1为h、含c
1~6
的烷基、含c
3~7
的环烷基、苯基或取代苯基;
[0010]
基团r2为h、含c
1~6
的烷基、含c
1~6
的卤代烷基、含c
3~7
的环烷基、含c
3~6
的卤代环烷基、苯基或取代苯基;
[0011]
基团r3为h、含c
1~6
的烷基、苯基或取代苯基、卤素、含c
1~6
的烷氧基、氰基、硝基、三氟甲基;
[0012]
基团r4为羟基、含c
1~6
的烷氧基、氨基或取代氨基、苯胺基、苄胺基或取代苄胺基、哌啶基、哌嗪基、n
‑
取代哌嗪基、n,n
‑
双取代氨基、c
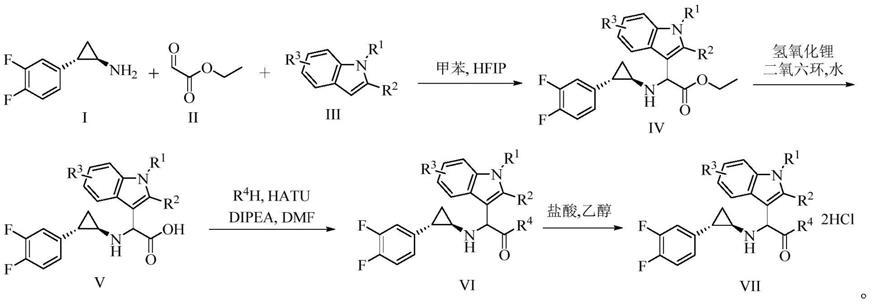
3~6
环状氨基。
[0013]
其中,本文中的“取代苯基”是指含c
1~6
的烷基取代的苯基、卤代苯基、氰基取代的苯基、硝基取代的苯基或氨基取代的苯基。本文中的“取代氨基”是指含c
1~6
的烷氨基或卤素取代的氨基。本文中的“取代苄胺基”是指苄胺基中的苯基上的碳原子被取代,具体是指卤素取代的苄氨基、含c
1~3
的烷基取代的苄氨基、含c
1~3
的烷氧基取代的苄氨基、氰基取代的苄氨基或硝基取代的苄氨基。
[0014]
从上述通式vi和通式vii来看,上述通式vii相当于通式vi的盐酸盐。即,所述3,4
‑
二氟苯环丙胺类衍生物为通式vi所示的化合物或其盐酸盐。
[0015]
优选地,所述基团r1为h、甲基、乙基、丙基、环丙基或苯基。
[0016]
优选地,所述基团r2为h、甲基、乙基、丙基、丁基、环丙基、环丁基、苯基、或卤苯基。其中,卤苯基为4
‑
氟苯基、4
‑
氯苯基或4
‑
溴苯基。
[0017]
优选地,所述基团r3为h、4
‑
甲基、4
‑
乙基、4
‑
丙基、4
‑
丁基、4
‑
氟、4
‑
溴、3
‑
苯基、3
‑
苄基、3
‑
甲氧基、3
‑
乙氧基、3
‑
氰基、3
‑
硝基、或4
‑
三氟甲基;其中,“4
‑”
是指基团r3所在的苯基的第4位,“3
‑”
是基团r3所在苯基的第3位。
[0018]
所述基团r4为羟基、甲氧基、乙氧基、戊氧基、氨基、氟代氨基、甲氨基、乙氨基、为羟基、甲氧基、乙氧基、戊氧基、氨基、氟代氨基、甲氨基、乙氨基、(n
‑
叔丁氧羰基哌嗪基)、叔丁氧羰基哌嗪基)、
[0019]
基于上述,所述3,4
‑
二氟苯环丙胺类衍生物为具有所述通式vi和下列基团的化合物vi
‑
1~vi
‑
20中的一个:
[0020]
vi
‑
1:r1=h,r2=h,r3=h,
[0021]
vi
‑
2:r1=h,r2=h,r3=h,
[0022]
vi
‑
3:r1=h,r2=h,r3=h,
[0023]
vi
‑
4:r1=h,r2=h,r3=h,
[0024]
vi
‑
5:r1=h,r2=h,r3=h,
[0025]
vi
‑
6:r1=h,r2=h,r3=h,
[0026]
vi
‑
7:r1=h,r2=h,r3=h,
[0027]
vi
‑
8:r1=h,r2=h,r3=h,
[0028]
vi
‑
9:r1=h,r2=h,r3=h,
[0029]
vi
‑
10:r1=h,r2=h,r3=h,
[0030]
vi
‑
11:r1=h,r2=h,r3=h,
[0031]
vi
‑
12:r1=h,r2=h,r3=h,
[0032]
vi
‑
13:r1=h,r2=h,r3=h,
[0033]
vi
‑
14:r1=h,r2=h,r3=h,
[0034]
vi
‑
15:r1=h,r2=h,r3=h,
[0035]
vi
‑
16:r1=h,r2=h,r3=h,
[0036]
vi
‑
17:r1=h,r2=h,r3=h,
[0037]
vi
‑
18:r1=h,r2=h,r3=h,
[0038]
vi
‑
19:r1=h,r2=h,r3=h,
[0039]
vi
‑
20:r1=h,r2=h,r3=h,
[0040]
基于上述,所述3,4
‑
二氟苯环丙胺类衍生物为具有所述通式vii和下列基团的化合物vi
‑
1~vi
‑
13中的一个:
[0041]
vii
‑
1:r1=h,r2=h,r3=h,
[0042]
vii
‑
2:r1=h,r2=h,r3=h,
[0043]
vii
‑
3:r1=h,r2=h,r3=h,
[0044]
vii
‑
4:r1=h,r2=h,r3=h,
[0045]
vii
‑
5:r1=ch3,r2=h,r3=h,
[0046]
vii
‑
6:r2=h,r3=h,
[0047]
vii
‑
7:r2=h,r3=h,
[0048]
vii
‑
8:r1=h,r2=ch3,r3=h,
[0049]
vii
‑
9:r1=h,r3=h,
[0050]
vii
‑
10:r1=h,r3=h,
[0051]
vii
‑
11:r1=h,r2=h,r3=4
‑
ch3,
[0052]
vii
‑
12:r1=h,r2=h,r3=4
‑
f,
[0053]
vii
‑
13:r1=h,r2=h,r3=4
‑
br,
[0054]
本发明还提供一种上述二氟苯环丙胺类化合物的制备方法,包括步骤:
[0055]
中间体iv的制备将3,4
‑
二氟苯环丙胺和乙醛酸乙酯溶于甲苯混合溶剂中,然后在常温下与吲哚化合物iii反应,制得中间体iv;其中,所述甲苯混合溶剂主要由甲苯和六氟
异丙醇(英文缩写“hfip”)组成,所述吲哚化合物iii的结构通式为:中间体iv的结构通式为:
[0056]
中间体v的制备所述中间体iv先在有机溶剂中与氢氧化锂水溶液在室温下反应;再去除所述有机溶剂,并用盐酸调节ph至3~4使析出固体;然后抽滤分离,制得中间体v;其中,所述有机溶剂为四氢呋喃或二氧六环,所述中间体v的结构通式为
[0057]
通式vi的制备常温下将所述中间体v和hatu(其中文名称:2
‑
(7
‑
氮杂苯并三氮唑)
‑
n,n,n',n'
‑
四甲基脲六氟磷酸酯)溶于dmf(中文名称:n,n
‑
二甲基甲酰胺)中,形成体系s1;将胺类化合物r4h和有机碱dipea(中文名称:n,n
‑
二异丙基乙胺)溶于dmf中,形成体系s2;将所述体系s2滴加到所述体系s1中并于室温下反应,制得产物通式vi。
[0058]
在中间体iv的制备的步骤中,所述甲苯混合溶剂中的hfip主要用于促进3,4
‑
二氟苯环丙胺与乙醛酸乙酯的缩合反应生成亚胺,以及后续与吲哚化合物iii的加成反应;甲苯主要用来增加反应底物溶解度。中间体iv的合成机理为:通过将3,4
‑
二氟苯环丙胺和乙醛酸乙酯在hfip和甲苯组成的甲苯混合溶剂中预活化形成亚胺中间体,加入吲哚化合物iii后通过hfip促进的加成反应直接将三分子合并成一个分子,以形成中间体iv。
[0059]
基于上述制备方法,所述中间体iv的制备的步骤包括:将3,4
‑
二氟苯环丙胺和乙醛酸乙酯按照1:1~1:3的摩尔比与所述混合溶剂在常温下搅拌均匀,其中所述混合溶剂中的甲苯与六氟异丙醇的体积比为1:1~5:1;然后再加入所述取代吲哚iii继续常温搅拌6~12h,经tlc监测反应完全后,冷却至室温,加入硅胶炒样,柱层析分离,得到所述中间体iv。
[0060]
基于上述制备方法,所述中间体v的制备的步骤包括:将所述中间体iv溶于二氧六环中,0℃下冰浴搅拌形成中间体iv混合液;随后将氢氧化锂溶于水中形成所述氢氧化锂水溶液,其中,所述中间体iv与氢氧化锂的摩尔比为1:1~1:10,二氧六环与水的体积比为1:1~20:1;将所述氢氧化锂水溶液滴加至所述中间体iv混合液中,继续室温搅拌反应4~6h;经tlc监测反应完全后,冷却至室温,减压蒸馏除去体系中的二氧六环,用稀盐酸溶液调节ph到3~4使固体析出;然后进行抽滤、冰水洗涤、烘干处理即可得到所述中间体v。
[0061]
基于上述制备方法,所述通式vi的制备的步骤包括:将所述中间体v和hatu按照1:1~1:3的摩尔比溶于dmf,常温搅拌1~48h,得到所述体系s1;同时将胺类化合物r4h和有机碱dipea按照1:1~1:3的摩尔比溶于dmf,常温搅拌0.5~2h,得到所述体系s2;将所述体系
s2滴加至所述体系s1中,并继续常温反应6~8h;经tlc监测反应完全后,形成产物混合物;向所述产物混合物中加入等量的水和乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并所述有机层;所述有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到所述通式vi的产物。
[0062]
基于上述制备方法,还包括步骤通式vii的制备,将所述通式vi溶于乙醇中并滴加浓盐酸于室温下反应,制得所述通式vi的盐酸盐产物通式vii。具体地,将所述中间体vi溶于乙醇中,常温搅拌,形成摩尔浓度为0.1~10mmol/ml的中间体vi溶液;向所述中间体vi溶液中滴加2~3滴浓盐酸并继续搅拌6~8h;经tlc监测反应完全后,减压蒸馏除去其中的溶剂,向残余物中加入乙酸乙酯使产品析出,抽滤,滤饼用乙酯洗涤,即可得到盐酸盐产物通式vii。
[0063]
所以,本发明提供的上述二氟苯环丙胺类化合物的反应历程如下:
[0064][0065]
本发明还提供一种上述二氟苯环丙胺类化合物在作为制备lsd1抑制剂中的应用。
[0066]
因此,本案提供的上述二氟苯环丙胺类化合物是一类3,4
‑
二氟苯环丙胺类衍生物或其药学可接受的盐,经试验验证,其具有抑制lsd1的作用,从而使得本发明为寻找一类新的基于lsd1靶点的抗肿瘤药物开辟一条新途径。
[0067]
另外,本发明还提供一种上述3,4
‑
二氟苯环丙胺类化合物的制备方法,该方法包括依次分别制备中间体iv、中间体v、中间体vi以及中间体vii,其中中间体iv采用三原料一锅反应法制备,通过将3,4
‑
二氟苯环丙胺和乙醛酸乙酯在hfip和甲苯的混合溶剂中预活化形成亚胺中间体,加入吲哚化合物iii后通过hfip促进的加成反应直接将三分子合并成一个分子,制得中间体iv,该制备方法无需过渡金属催化,也无需高温加热和惰性气体保护,制备方法简单,简化了制备3,4
‑
二氟苯环丙胺类化合物的步骤;同时,底物适用范围广,产率高,可高达85%。
具体实施方式
[0068]
下面通过具体实施方式,对本发明的技术方案做进一步的详细描述。若未特别指明,以下实施例中所用的技术手段为本领域技术人员所熟知的常规手段。
[0069]
实施例1
[0070]
本实施例提供一种化合物vi
‑
1,该化合物的结构式如下:
[0071][0072]
化合物vi
‑
1的合成历程如下所示:
[0073][0074]
具体地,本实施例还提供一种化合物vi
‑
1制备方法,包括以下步骤:
[0075]
中间体iv
‑
1的制备:将2.53g、约15mmol的原料3,4
‑
二氟苯环丙胺i和1.02g、约10mmol的乙醛酸乙酯ii加入至圆底烧瓶中,随后向其中加入混合溶剂,且该混合溶剂有12ml甲苯和4ml六氟异丙醇组成;常温搅拌约30min后,再向反应体系中加入1.17g、约10mmol的吲哚iii
‑
1继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到2.5g黄色油状物iv
‑
1,产率为68%。
[0076]
中间体v
‑
1的制备:将3.7g、约10mmol的中间体iv
‑
1溶于15ml二氧六环中,0℃下冰浴搅拌,随后将957mg、约40mmol氢氧化锂溶于10ml水中,滴加至反应体系中,继续室温搅拌反应6h。经tlc监测反应完全后,体系冷却至室温,减压蒸馏除去体系中的二氧六环,用1m的稀盐酸溶液调节ph到3~4使固体析出。将反应体系抽滤,滤饼用少量冰水洗涤,烘干即可得到3.4g白色固体v
‑
1,产率为99%。
[0077]
产物vi
‑
1的制备:将342mg、约1mmol的中间体v
‑
1和574mg、约1.5mmol的hatu溶于dmf,常温搅拌1h,得到体系s1,同时将140mg、约1.5mmol苯胺和195mg、约1.5mmol的有机碱dipea溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应8h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到50mg黄色固体vi
‑
1,产率为12%;1h nmr(400mhz,dmso
‑
d6)δ11.03
‑
10.93(m,1h),10.10
‑
9.97(m,1h),7.82
‑
7.68(m,1h),7.67
‑
7.57(m,2h),7.39
‑
7.15(m,5h),7.12
‑
6.92(m,4h),6.90
‑
6.76(m,1h),4.75(s,1h),3.25(s,1h),2.32
‑
2.20(m,1h),2.05
‑
1.90(m,1h),1.21
‑
1.11(m,1h),1.10
‑
0.98(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
1的结构式如上所示。
[0078]
实施例2
[0079]
本实施例提供一种化合物vi
‑
2,该化合物的结构式如下:
[0080][0081]
本实施例还提供一种化合物vi
‑
2制备方法,包括以下步骤:
[0082]
产物vi
‑
2的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将160mg苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应8h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到57mg黄色固体vi
‑
2,产率为13%;1h nmr(400mhz,dmso
‑
d6)δ10.97
‑
10.87(m,1h),8.61
‑
8.48(m,1h),7.77
‑
7.61(m,1h),7.33(t,j=7.5hz,1h),7.28
‑
7.15(m,6h),7.13
‑
7.01(m,2h),7.01
‑
6.89(m,2h),6.85
‑
6.77(m,1h),4.61(d,j=5.4hz,1h),4.38
‑
4.14(m,2h),3.05(s,1h),2.28
‑
2.17(m,1h),2.03
‑
1.85(m,1h),1.14
‑
1.05(m,1h),1.04
‑
0.94(t,j=5.8hz,1h);因此,可以确定本实施例提供的所述化合物vi
‑
2的结构式如上所示。
[0083]
实施例3
[0084]
本实施例提供一种化合物vi
‑
3,该化合物的结构式如下:
[0085][0086]
本实施例还提供一种化合物vi
‑
3制备方法,包括以下步骤:
[0087]
产物vi
‑
3的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将181mg对甲基苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应8h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到49mg黄色固体vi
‑
3,产率为11%;1h nmr(400mhz,dmso
‑
d6)δ10.92(d,j=7.5hz,1h),8.57
‑
8.44(m,1h),8.06(t,j=5.4hz,1h),7.77
‑
7.60(m,1h),7.33(t,j=7.4hz,1h),7.27
‑
7.24(m,2h),7.22
‑
7.19(m,2h),7.10(d,j=7.8hz,1h),7.06
‑
7.03(m,1h),6.99(s,1h),6.96
‑
6.90(m,1h),6.86
‑
6.76(m,1h),4.60(d,j=7.6hz,1h),4.33
‑
4.26(m,2h),3.05(s,1h),2.30(s,2h),2.26
‑
2.22(m,2h),2.03
‑
1.87(m,1h),1.14
‑
1.06(m,1h),1.04
‑
0.94(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
3的结构式如上所示。
[0088]
实施例4
[0089]
本实施例提供一种化合物vi
‑
4,该化合物的结构式如下:
[0090][0091]
本实施例还提供一种化合物vi
‑
4制备方法,包括以下步骤:
[0092]
产物vi
‑
4的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将205mg对甲氧基苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到48mg黄色固体vi
‑
4,产率为10%;1h nmr(400mhz,dmso
‑
d6)δ10.91(d,j=7.2hz,1h),8.53
‑
8.43(m,1h),7.77
‑
7.59(m,1h),7.38
‑
7.26(m,2h),7.25
‑
7.18(m,1h),7.18
‑
7.10(m,2h),7.09
‑
7.01(m,2h),7.00
‑
6.89(m,2h),6.84
‑
6.70(m,1h),4.58(d,j=8.3hz,1h),4.32
‑
4.20(m,2h),3.75(s,1h),3.70(s,3h),3.04(s,1h),2.27
‑
2.14(m,1h),2.09
‑
1.81(m,1h),1.14
‑
1.05(m,1h),1.03
‑
0.93(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
4的结构式如上所示。
[0093]
实施例5
[0094]
本实施例提供一种化合物vi
‑
5,该化合物的结构式如下:
[0095][0096]
本实施例还提供一种化合物vi
‑
5制备方法,包括以下步骤:
[0097]
产物vi
‑
5的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将275mg二苯甲胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到55mg黄色固体vi
‑
5,产率为11%;1h nmr(400mhz,dmso
‑
d6)δ10.89(s,1h),8.95(t,j=7.9hz,1h),7.76
‑
7.61(m,1h),7.47
‑
7.34(m,2h),7.33
‑
7.27(m,3h),7.25
‑
7.18(m,5h),7.17
‑
7.09(m,3h),7.05(t,j=7.6hz,1h),6.99
‑
6.64(m,3h),6.23
‑
6.09(m,1h),4.81
‑
4.70(m,1h),3.11(s,1h),2.25
‑
2.15(m,1h),2.01
‑
1.88(m,1h),1.15
‑
1.03(m,1h),1.03
‑
0.93(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
5的结构式如上所示。
[0098]
实施例6
[0099]
本实施例提供一种化合物vi
‑
6,该化合物的结构式如下:
[0100][0101]
本实施例还提供一种化合物vi
‑
6制备方法,包括以下步骤:
[0102]
产物vi
‑
6的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将198mg对氰基苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到57mg黄色固体vi
‑
6,产率为13%;1h nmr(400mhz,dmso
‑
d6)δ10.98(d,j=7.5hz,1h),8.71(t,j=6.1hz,1h),7.84
‑
7.79(m,1h),7.68(d,j=8.2hz,1h),7.64(d,j=7.9hz,1h),7.54
‑
7.49(m,1h),7.38(d,j=8.0hz,2h),7.34(d,j=8.0hz,1h),7.26
‑
7.22(m,1h),7.18(d,j=2.4hz,1h),7.09
‑
7.04(m,1h),6.96
‑
6.91(m,1h),6.84
‑
6.77(m,1h),4.66(s,1h),4.41
‑
4.36(m,2h),2.89(s,1h),2.25
‑
2.17(m,1h),2.05
‑
1.95(m,1h),1.14
‑
1.07(m,1h),1.04
‑
0.97(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
6的结构式如上所示。
[0103]
实施例7
[0104]
本实施例提供一种化合物vi
‑
7,该化合物的结构式如下:
[0105][0106]
本实施例还提供一种化合物vi
‑
7制备方法,包括以下步骤:
[0107]
产物vi
‑
7的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将187mg对氟苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到62mg黄色固体vi
‑
7,产率为14%;1h nmr(400mhz,dmso
‑
d6)δ10.82(d,j=7.5hz,1h),8.61
‑
8.53(m,1h),8.07(s,1h),7.75
‑
7.59(m,1h),7.46
‑
7.40(m,2h),7.33(t,j=7.7hz,1h),7.26
‑
7.24(m,1h),7.22
‑
7.19(m,1h),7.17
‑
7.13(m,1h),7.07
‑
7.04(m,1h),7.01
‑
6.94(m,2h),6.85
‑
6.75(m,1h),4.59(d,j=5.3hz,1h),4.34(d,j=4.3hz,2h),2.89(s,1h),2.24
‑
2.15(m,1h),2.00
‑
1.85(m,1h),1.13
‑
1.06(m,1h),1.03
‑
0.95(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
7的结构式如上所示。
[0108]
实施例8
[0109]
本实施例提供一种化合物vi
‑
8,该化合物的结构式如下:
[0110][0111]
本实施例还提供一种化合物vi
‑
8制备方法,包括以下步骤:
[0112]
产物vi
‑
8的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将181mg n
‑
甲基苄胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到61mg黄色固体vi
‑
8,产率为14%;1h nmr(400mhz,dmso
‑
d6)δ10.99(d,j=7.5hz,1h),7.80
‑
7.51(m,1h),7.39
‑
7.33(m,1h),7.33
‑
7.26(m,1h),7.25
‑
7.20(m,2h),7.18
‑
7.11(m,2h),7.11
‑
7.02(m,2h),6.99(t,j=7.6hz,2h),6.92(d,j=7.4hz,1h),6.87
‑
6.75(m,1h),5.10
‑
4.88(m,1h),4.77
‑
4.56(m,1h),4.53
‑
4.05(m,1h),3.04(s,1h),2.86
‑
2.75(m,3h),2.30
‑
2.09(m,1h),2.02
‑
1.80(m,1h),1.15
‑
0.88(m,2h);因此,可以确定本实施例提供的所述化合物vi
‑
8的结构式如上所示。
[0113]
实施例9
[0114]
本实施例提供一种化合物vi
‑
9,该化合物的结构式如下:
[0115][0116]
本实施例还提供一种化合物vi
‑
9制备方法,包括以下步骤:
[0117]
产物vi
‑
9的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将150mg n
‑
甲基哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到76mg黄色固体vi
‑
9,产率为18%;1h nmr(400mhz,dmso
‑
d6)δ11.00
‑
10.97(m,1h),7.75
‑
7.57(m,1h),7.33(t,j=9.2hz,1h),7.28
‑
7.21(m,1h),7.21
‑
7.11(m,1h),7.10
‑
7.01(m,2h),7.00
‑
6.88(m,1h),6.87
‑
6.77(m,1h),5.08
‑
4.98(m,1h),3.69
‑
3.42(m,3h),3.29
‑
3.00(m,2h),2.29
‑
2.08(m,3h),2.07
‑
1.97(m,3h),1.97
‑
1.75(m,3h),1.18
‑
1.04(m,1h),1.03
‑
0.94(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
9的结构式如上所示。
[0118]
实施例10
[0119]
本实施例提供一种化合物vi
‑
10,该化合物的结构式如下:
[0120][0121]
本实施例还提供一种化合物vi
‑
10制备方法,包括以下步骤:
[0122]
产物vi
‑
10的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将171mg n
‑
乙基哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到66mg黄色固体vi
‑
10,产率为15%;1h nmr(400mhz,dmso
‑
d6)δ10.99
‑
10.89(m,1h),7.74
‑
7.55(m,1h),7.33(t,j=9.2hz,1h),7.25(t,j=9.3hz,1h),7.19
‑
7.09(m,1h),7.09
‑
6.99(m,2h),6.99
‑
6.87(m,1h),6.87
‑
6.79(m,1h),5.07
‑
4.94(m,1h),3.75
‑
3.38(m,3h),3.29
‑
3.08(m,2h),2.95(s,1h),2.36
‑
2.24(m,1h),2.20
‑
2.07(m,3h),2.04
‑
1.88(m,2h),1.87
‑
1.74(m,1h),1.12
‑
1.02(m,1h),1.02
‑
0.95(m,1h),0.93
‑
0.81(m,3h);因此,可以确定本实施例提供的所述化合物vi
‑
10的结构式如上所示。
[0123]
实施例11
[0124]
本实施例提供一种化合物vi
‑
11,该化合物的结构式如下:
[0125][0126]
本实施例还提供一种化合物vi
‑
11制备方法,包括以下步骤:
[0127]
产物vi
‑
11的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将279mg n
‑
boc哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到102mg黄色固体vi
‑
11,产率为20%;1h nmr(400mhz,dmso
‑
d6)δ11.01
‑
10.91(m,1h),7.73
‑
7.57(m,1h),7.33(t,j=9.4hz,1h),7.28
‑
7.11(m,2h),7.10
‑
6.99(m,2h),6.98
‑
6.89(m,1h),6.88
‑
6.74(m,1h),5.02(s,1h),3.55
‑
3.46(m,2h),3.28
‑
3.08(m,3h),2.93
‑
2.84(m,4h),2.14(s,1h),1.91(s,1h),1.42(s,3h),1.34(s,6h),1.16
‑
1.03(m,1h),1.03
‑
0.91(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
11的结构式如上所示。
[0128]
实施例12
[0129]
本实施例提供一种化合物vi
‑
12,该化合物的结构式如下:
[0130][0131]
本实施例还提供一种化合物vi
‑
12制备方法,包括以下步骤:
[0132]
产物vi
‑
12的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将192mg n
‑
乙酰基哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到44mg黄色固体vi
‑
12,产率为10%;1h nmr(400mhz,dmso
‑
d6)δ11.03
‑
10.92(m,1h),7.64
‑
7.56(m,1h),7.36
‑
7.30(m,1h),7.27
‑
7.20(m,1h),7.18
‑
7.11(m,1h),7.09
‑
7.00(m,2h),6.91(t,j=7.6hz,1h),6.88
‑
6.76(m,1h),5.04(s,1h),3.70
‑
3.46(m,4h),3.42
‑
3.37(m,1h),3.30
‑
3.19(m,3h),2.13(s,1h),2.00
‑
1.82(m,5h),1.12
‑
1.06(m,1h),1.03
‑
0.94(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
12的结构式如上所示。
[0133]
实施例13
[0134]
本实施例提供一种化合物vi
‑
13,该化合物的结构式如下:
[0135][0136]
本实施例还提供一种化合物vi
‑
13制备方法,包括以下步骤:
[0137]
产物vi
‑
13的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将288mg 1
‑
(4
‑
甲氧基苯基)哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到43mg黄色固体vi
‑
13,产率为8%;1h nmr(400mhz,dmso
‑
d6)δ11.00
‑
10.90(m,1h),7.76
‑
7.58(m,1h),7.37
‑
7.28(m,1h),7.25
‑
7.10(m,2h),7.09
‑
6.99(m,2h),6.99
‑
6.88(m,1h),6.87
‑
6.80(m,1h),6.79
‑
6.70(m,4h),5.14
‑
5.00(m,1h),3.80
‑
3.56(m,6h),3.53
‑
3.38(m,1h),3.00
‑
2.67(m,5h),2.23
‑
2.10(m,1h),2.03
‑
1.88(m,1h),1.18
‑
1.04(m,1h),1.02
‑
0.94(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
13的结构式如上所示。
[0138]
实施例14
[0139]
本实施例提供一种化合物vi
‑
14,该化合物的结构式如下:
[0140][0141]
本实施例还提供一种化合物vi
‑
14制备方法,包括以下步骤:
[0142]
产物vi
‑
14的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将195mg 1
‑
(2
‑
羟乙基)哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到55mg黄色固体vi
‑
14,产率为12%;1h nmr(400mhz,dmso
‑
d6)δ11.00
‑
10.91(m,1h),7.73
‑
7.56(m,1h),7.37
‑
7.30(m,1h),7.28
‑
7.21(m,1h),7.20
‑
7.10(m,1h),7.10
‑
7.01(m,2h),7.00
‑
6.87(m,1h),6.86
‑
6.78(m,1h),5.07
‑
4.98(m,1h),4.41
‑
4.33(m,1h),3.67
‑
3.40(m,5h),3.28
‑
3.11(m,2h),2.35
‑
2.09(m,6h),1.96
‑
1.84(m,2h),1.13
‑
1.04(m,1h),1.03
‑
0.95(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
14的结构式如上所示。
[0143]
实施例15
[0144]
本实施例提供一种化合物vi
‑
15,该化合物的结构式如下:
[0145][0146]
本实施例还提供一种化合物vi
‑
15制备方法,包括以下步骤:
[0147]
产物vi
‑
15的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将348mg 1
‑
(4
‑
叔丁基苄基)哌嗪(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到51mg黄色固体vi
‑
15,产率为9%;1h nmr(400mhz,dmso
‑
d6)δ10.99
‑
10.92(m,1h),7.73
‑
7.57(m,1h),7.34(t,j=8.5hz,1h),7.30(d,j=8.1hz,2h),7.27
‑
7.21(m,1h),7.20
‑
7.14(m,1h),7.14
‑
7.09(m,2h),7.08
‑
7.01(m,2h),6.99
‑
6.88(m,1h),6.86
‑
6.79(m,1h),5.07
‑
4.99(m,1h),3.74
‑
3.54(m,1h),3.54
‑
3.43(m,2h),3.32
‑
3.24(m,2h),3.23
‑
3.11(m,2h),2.33
‑
2.24(m,1h),2.21
‑
2.09(m,2h),2.04
‑
1.90(m,2h),1.90
‑
1.72(m,1h),1.24(s,9h),1.13
‑
1.04(m,1h),1.03
‑
0.93(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
15的结构式如上所示。
[0148]
实施例16
[0149]
本实施例提供一种化合物vi
‑
16,该化合物的结构式如下:
[0150][0151]
本实施例还提供一种化合物vi
‑
16制备方法,包括以下步骤:
[0152]
产物vi
‑
16的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将149mg 4
‑
哌啶酮(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到45mg黄色固体vi
‑
16,产率为11%;1h nmr(400mhz,dmso
‑
d6)δ10.95
‑
10.86(m,1h),8.15
‑
8.07(m,1h),7.76
‑
7.61(m,1h),7.37
‑
7.29(m,1h),7.28
‑
7.22(m,1h),7.22
‑
7.15(m,1h),7.10
‑
7.00(m,2h),7.00
‑
6.89(m,1h),6.89
‑
6.82(m,1h),4.61
‑
4.53(m,1h),4.15
‑
4.00(m,1h),3.03(s,1h),2.45
‑
2.29(m,2h),2.27
‑
2.12(m,3h),2.03
‑
1.84(m,3h),1.73
‑
1.55(m,2h),1.16
‑
1.07(m,1h),1.07
‑
0.96(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
16的结构式如上所示。
[0153]
实施例17
[0154]
本实施例提供一种化合物vi
‑
17,该化合物的结构式如下:
[0155][0156]
本实施例还提供一种化合物vi
‑
17制备方法,包括以下步骤:
[0157]
产物vi
‑
17的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将106mg环丙甲胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到46mg黄色固体vi
‑
17,产率为12%;1h nmr(400mhz,dmso
‑
d6)δ10.94
‑
10.85(m,1h),8.17
‑
8.05(m,1h),7.74
‑
7.62(m,1h),7.37
‑
7.23(m,2h),7.23
‑
7.14(m,1h),7.11
‑
6.99(m,2h),6.99
‑
6.89(m,1h),6.88
‑
6.79(m,1h),4.53(s,1h),3.11
‑
2.95(m,2h),2.94
‑
2.84(m,1h),2.21(s,1h),2.05
‑
1.84(m,1h),1.18
‑
0.96(m,3h),0.94
‑
0.78(m,1h),0.54
‑
0.26(m,2h),0.25
‑
0.05(m,2h);因此,可以确定本实施例提供的所述化合物vi
‑
17的结构式如上所示。
[0158]
实施例18
[0159]
本实施例提供一种化合物vi
‑
18,该化合物的结构式如下:
[0160][0161]
本实施例还提供一种化合物vi
‑
18制备方法,包括以下步骤:
[0162]
产物vi
‑
18的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将176mg 3
‑
异丙氧基丙胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到47mg黄色固体vi
‑
18,产率为11%;1h nmr(400mhz,dmso
‑
d6)δ10.95
‑
10.87(m,1h),8.06
‑
7.98(m,1h),7.75
‑
7.61(m,1h),7.33(t,j=7.6hz,1h),7.29
‑
7.21(m,1h),7.20
‑
7.15(m,1h),7.11
‑
6.99(m,2h),6.99
‑
6.90(m,1h),6.89
‑
6.78(m,1h),4.51(s,1h),3.45
‑
3.36(m,1h),3.31
‑
3.19(m,2h),3.18
‑
3.08(m,2h),2.24
‑
2.16(m,1h),2.04
‑
1.84(m,1h),1.65
‑
1.48(m,2h),1.14
‑
0.92(m,9h);因此,可以确定本实施例提供的所述化合物vi
‑
18的结构式如上所示。
[0163]
实施例19
[0164]
本实施例提供一种化合物vi
‑
19,该化合物的结构式如下:
[0165][0166]
本实施例还提供一种化合物vi
‑
19制备方法,包括以下步骤:
[0167]
产物vi
‑
19的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将153mg n,n
‑
二甲基丙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到64mg黄色固体vi
‑
19,产率为15%;1h nmr(400mhz,dmso
‑
d6)δ10.98
‑
10.89(m,1h),8.08(t,j=5.6hz,1h),7.64(d,j=8.0hz,1h),7.35
‑
7.30(m,1h),7.28
‑
7.22(m,1h),7.18
‑
7.14(m,1h),7.07
‑
7.02(m,1h),7.02
‑
6.95(m,1h),6.93(t,j=7.5hz,1h),6.89
‑
6.83(m,1h),4.50(s,1h),3.15
‑
3.04(m,2h),2.23
‑
2.07(m,5h),2.06
‑
2.00(m,6h),1.56
‑
1.44(m,2h),1.13
‑
1.05(m,1h),1.04
‑
0.95(m,1h);因此,可以确定本实施例提供的所述化合物vi
‑
19的结构式如上所示。
[0168]
实施例20
[0169]
本实施例提供一种化合物vi
‑
20,该化合物的结构式如下:
[0170][0171]
本实施例还提供一种化合物vi
‑
20制备方法,包括以下步骤:
[0172]
产物vi
‑
20的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将110mg二乙胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到40mg黄色固体vi
‑
20,产率为10%;1h nmr(400mhz,dmso
‑
d6)δ11.00
‑
10.91(m,1h),7.76
‑
7.59(m,1h),7.36
‑
7.31(m,1h),7.28
‑
7.21(m,1h),7.16
‑
7.10(m,1h),7.08
‑
7.06(m,1h),7.04
‑
6.99(m,1h),6.98
‑
6.88(m,1h),6.88
‑
6.80(m,1h),4.87(s,1h),3.52
‑
3.44(m,1h),3.31
‑
3.24(m,1h),3.23
‑
3.05(m,3h),2.19
‑
2.10(m,1h),2.00
‑
1.91(m,1h),1.03
‑
0.97(m,3h),0.96
‑
0.90(m,3h),0.81
‑
0.68(m,2h);因此,可以确定本实施例提供的所述化合物vi
‑
20的结构式如上所示。
[0173]
实施例21
[0174]
本实施例提供一种化合物vii
‑
1,该化合物的结构式如下:
[0175][0176]
本实施例还提供一种化合物vii
‑
1的制备方法,包括以下步骤:
[0177]
产物vii
‑
1的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将192mg 4
‑
二甲氨基哌啶(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到66mg黄色油状物,产率为15%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
1;1h nmr(400mhz,dmso
‑
d6)δ11.87
‑
11.66(m,1h),11.42
‑
10.98(m,1h),10.35
‑
9.33(m,2h),7.83
‑
7.68(m,1h),7.65
‑
7.53(m,1h),7.52
‑
7.40(m,1h),7.38
‑
7.27(m,1h),7.27
‑
7.17(m,1h),7.17
‑
7.05(m,1h),7.05
‑
6.97(m,1h),6.96
‑
6.54(m,1h),6.07(s,1h),4.65
‑
4.49(m,1h),4.20
‑
3.90(m,2h),3.37
‑
3.25(m,1h),3.15
‑
2.96(m,1h),2.81
‑
2.56(m,6h),2.32
‑
2.02(m,4h),2.01
‑
1.80(m,1h),1.73
‑
1.51(m,1h),1.48
‑
1.26(m,1h),1.24
‑
1.01(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
1的结构式如上所示。
[0178]
实施例22
[0179]
本实施例提供一种化合物vii
‑
2,该化合物的结构式如下:
[0180][0181]
本实施例还提供一种化合物vii
‑
2的制备方法,包括以下步骤:
[0182]
产物vii
‑
2的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到36mg黄色油状物,产率为9%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
2;1h nmr(400mhz,dmso
‑
d6)δ11.58
‑
11.50(,1h),10.39(s,1h),10.35
‑
9.57(m,2h),9.04
‑
8.94(m,1h),7.80
‑
7.57(m,1h),7.56
‑
7.45(m,1h),7.45
‑
7.28(m,2h),7.21
‑
7.07(m,2h),7.05
‑
6.92(m,1h),6.92
‑
6.56(m,1h),5.55
‑
5.33(m,1h),3.75
‑
3.63(m,1h),3.39(s,2h),3.31
‑
3.20(m,2h),3.11(s,1h),2.81
‑
2.61(m,6h),1.66
‑
1.42(m,1h),1.34
‑
1.21(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
2的结构式如上所示。
[0183]
实施例23
[0184]
本实施例提供一种化合物vii
‑
3,该化合物的结构式如下:
[0185][0186]
本实施例还提供一种化合物vii
‑
3的制备方法,包括以下步骤:
[0187]
产物vii
‑
3的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将153mg n,n,n
’‑
三甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到40mg黄色油状物,产率为9%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
3;1h nmr(400mhz,dmso
‑
d6)δ11.66
‑
11.56(m,1h),10.37
‑
10.00(m,1h),10.00
‑
9.78(m,1h),9.77
‑
9.53(m,1h),7.80
‑
7.60(m,1h),7.60
‑
7.49(m,1h),7.47
‑
7.38(m,1h),7.38
‑
7.25(m,1h),7.23
‑
7.09(m,2h),7.07
‑
6.90(m,2h),5.78(s,1h),4.32
‑
4.11(m,1h),3.25
‑
3.13(m,2h),2.95
‑
2.89(m,3h),2.88
‑
2.82(m,3h),2.81
‑
2.74(m,3h),2.70
‑
2.56(m,3h),1.44
‑
1.25(m,1h),1.22
‑
1.14(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
3的结构式如上所示。
[0188]
实施例24
[0189]
本实施例提供一种化合物vii
‑
4,该化合物的结构式如下:
[0190][0191]
本实施例还提供一种化合物vii
‑
4的制备方法,包括以下步骤:
[0192]
产物vii
‑
4的制备:将342mg中间体v
‑
1(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将90mg乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到38mg黄色油状物,产率为10%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
4;1h nmr(400mhz,dmso
‑
d6)δ11.56
‑
11.48(m,1h),10.43
‑
10.13(m,1h),9.94
‑
9.57(m,1h),8.94(t,j=5.7hz,1h),8.18(s,3h),7.80
‑
7.58(m,1h),7.56
‑
7.45(m,1h),7.45
‑
7.26(m,2h),7.22
‑
7.08(m,2h),7.06
‑
6.94(m,1h),6.94
‑
6.54(m,1h),5.49
‑
5.29(m,1h),3.61
‑
3.52(m,1h),3.20
‑
3.09(m,1h),2.98
‑
2.88(m,1h),2.87
‑
2.76(m,1h),2.66
‑
2.20(m,2h),1.64
‑
1.44(m,1h),1.36
‑
1.17(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
4的结构式如上所示。
[0193]
实施例25
[0194]
本实施例提供一种化合物vii
‑
5,该化合物的结构式如下:
[0195][0196]
本实施例还提供一种化合物vii
‑
5的制备方法,包括以下步骤:
[0197]
中间体iv
‑
2的制备:本实施例提供的中间体iv
‑
2的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.31g、约10mmol的吲哚化合物iii
‑
2继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.26g黄色油状物iv
‑
2,产率为85%。其中,吲哚化合物iii
‑
2的结构式为中间体iv
‑
2的结构式为
[0198]
中间体v
‑
2的制备:本实施例提供的中间体iiv
‑
2的制备方法与实施例1提供的中
间体v
‑
1的制备方法基本相同,主要不同之处在于:采用3.84g、约10mmol的中间体iv
‑
2参加反应,得到3.55g白色固体v
‑
2,产率为99%。其中,中间体v
‑
2的结构式为
[0199]
产物vii
‑
5的制备:将356mg中间体v
‑
2(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到47mg黄色油状物,产率为11%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
5;1h nmr(400mhz,dmso
‑
d6)δ10.37(s,1h),10.36
‑
9.64(m,2h),9.07
‑
8.94(m,1h),7.80
‑
7.56(m,1h),7.52
‑
7.43(m,2h),7.39
‑
7.28(m,1h),7.24
‑
7.17(m,1h),7.16
‑
7.05(m,1h),6.99
‑
6.49(m,2h),5.58
‑
5.35(m,1h),3.79
‑
3.72(m,3h),3.71
‑
3.60(m,1h),3.32
‑
3.17(m,2h),3.16
‑
3.06(m,1h),2.82
‑
2.70(m,6h),2.65
‑
2.10(m,2h),1.65
‑
1.43(m,1h),1.34
‑
1.21(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
5的结构式如上所示。
[0200]
实施例26
[0201]
本实施例提供一种化合物vii
‑
6,该化合物的结构式如下:
[0202][0203]
本实施例还提供一种化合物vii
‑
6的制备方法,包括以下步骤:
[0204]
中间体iv
‑
3的制备:本实施例提供的中间体iv
‑
3的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.57g、约10mmol的吲哚化合物iii
‑
3继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.28g黄色油状物iv
‑
3,产率为80%。其中,吲哚化合物iii
‑
3的结构式为中间体iv
‑
3的结构式为
[0205]
中间体v
‑
3的制备:本实施例提供的中间体iv
‑
3的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用4.1g、约10mmol的中间体iv
‑
3参加反应,得到3.82g白色固体v
‑
3,产率为100%。其中,中间体v
‑
3的结构式
[0206]
产物vii
‑
6的制备:将382mg中间体v
‑
3(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到46mg黄色油状物,产率为10%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
6;1h nmr(400mhz,dmso
‑
d6)δ10.59
‑
10.16(m,2h),10.04
‑
9.56(m,1h),9.09
‑
8.99(m,1h),7.82
‑
7.63(m,1h),7.58
‑
7.52(m,1h),7.52
‑
7.41(m,1h),7.36
‑
7.11(m,3h),7.11
‑
6.96(m,1h),6.71
‑
6.42(m,1h),5.56
‑
5.30(m,1h),3.74
‑
3.61(m,1h),3.43
‑
3.32(m,1h),3.31
‑
3.16(m,2h),3.15
‑
3.05(m,1h),2.87
‑
2.62(m,7h),2.62
‑
2.53(m,1h),1.63
‑
1.47(m,1h),1.30
‑
1.22(m,1h),1.11
‑
0.99(m,2h),0.84
‑
0.65(m,2h);因此,可以确定本实施例提供的所述化合物vii
‑
6的结构式如上所示。
[0207]
实施例27
[0208]
本实施例提供一种化合物vii
‑
7,该化合物的结构式如下:
[0209][0210]
本实施例还提供一种化合物vii
‑
7的制备方法,包括以下步骤:
[0211]
中间体iv
‑
4的制备:本实施例提供的中间体iv
‑
4的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.93g、约10mmol的吲哚化合物iii
‑
4继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.35g黄色油状物iv
‑
4,产率为75%。其中,吲哚化合物iii
‑
4的结构式为
中间体iv
‑
4的结构式为
[0212]
中间体v
‑
4的制备:本实施例提供的中间体iv
‑
4的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用4.46g、约10mmol的中间体iv
‑
4参加反应,得到3.89g白色固体v
‑
4,产率为93%。其中,中间体v
‑
4的结构式
[0213]
产物vii
‑
7的制备:将418mg中间体v
‑
4(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到52mg黄色油状物,产率为11%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
7;1h nmr(400mhz,dmso
‑
d6)δ10.66
‑
10.26(m,2h),10.23
‑
9.63(m,1h),9.12
‑
9.02(m,1h),7.92
‑
7.80(m,1h),7.79
‑
7.69(m,1h),7.63
‑
7.50(m,3h),7.50
‑
7.38(m,3h),7.36
‑
7.27(m,1h),7.27
‑
7.19(m,1h),7.16
‑
6.94(m,2h),6.64
‑
6.40(m,1h),5.70
‑
5.44(m,1h),3.76
‑
3.65(m,1h),3.29
‑
3.17(m,2h),3.16
‑
3.06(m,1h),2.81
‑
2.70(m,7h),2.65
‑
2.04(m,1h),1.65
‑
1.51(m,1h),1.35
‑
1.21(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
7的结构式如上所示。
[0214]
实施例28
[0215]
本实施例提供一种化合物vii
‑
8,该化合物的结构式如下:
[0216][0217]
本实施例还提供一种化合物vii
‑
8的制备方法,包括以下步骤:
[0218]
中间体iv
‑
5的制备:本实施例提供的中间体iv
‑
5的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.31g、约10mmol的吲
哚化合物iii
‑
5继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到2.96g黄色油状物iv
‑
5,产率为77%。其中,吲哚化合物iii
‑
5的结构式为中间体iv
‑
5的结构式为
[0219]
中间体v
‑
5的制备:本实施例提供的中间体iv
‑
5的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用3.84g、约10mmol的中间体iv
‑
5参加反应,得到3.42g白色固体v
‑
5,产率为96%。其中,中间体v
‑
5的结构式
[0220]
产物vii
‑
8的制备:将356mg中间体v
‑
5(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到55mg黄色油状物,产率为13%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
8;1h nmr(400mhz,dmso
‑
d6)δ11.49
‑
11.40(m,1h),10.75
‑
10.47(m,1h),10.20
‑
9.80(m,1h),9.76
‑
9.43(m,1h),7.66
‑
7.52(m,1h),7.40
‑
7.31(m,1h),7.30
‑
7.23(m,1h),7.22
‑
7.15(m,1h),7.11
‑
6.99(m,2h),6.99
‑
6.89(m,1h),6.87
‑
6.58(m,1h),5.47
‑
5.27(m,1h),3.76
‑
3.61(m,1h),3.29
‑
3.14(m,1h),3.11
‑
3.02(m,1h),2.75
‑
2.60(m,8h),2.46
‑
2.17(m,3h),1.55
‑
1.41(m,1h),1.28
‑
1.16(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
8的结构式如上所示。
[0221]
实施例29
[0222]
本实施例提供一种化合物vii
‑
9,该化合物的结构式如下:
[0223][0224]
本实施例还提供一种化合物vii
‑
9的制备方法,包括以下步骤:
[0225]
中间体iv
‑
6的制备:本实施例提供的中间体iv
‑
6的制备方法与实施例1提供的中
间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.93g、约10mmol的吲哚化合物iii
‑
6继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.7g黄色油状物iv
‑
6,产率为83%。其中,吲哚化合物iii
‑
6的结构式为中间体iv
‑
6的结构式为
[0226]
中间体v
‑
6的制备:本实施例提供的中间体iv
‑
6的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用4.46g、约10mmol的中间体iv
‑
6参加反应,得到3.93g白色固体v
‑
6,产率为94%。其中,中间体v
‑
6的结构式
[0227]
产物vii
‑
29的制备:将418mg中间体v
‑
6(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到60mg黄色油状物,产率为12%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
9;1h nmr(400mhz,dmso
‑
d6)δ12.01
‑
11.86(m,1h),10.82
‑
10.56(m,1h),10.26
‑
9.85(m,1h),9.73
‑
9.46(m,1h),8.72
‑
8.56(m,1h),7.77
‑
7.69(m,1h),7.67
‑
7.58(m,2h),7.56
‑
7.50(m,1h),7.48
‑
7.43(m,1h),7.42
‑
7.34(m,2h),7.27
‑
7.16(m,2h),7.14
‑
7.03(m,1h),6.94
‑
6.70(m,1h),6.70
‑
6.38(m,1h),5.53
‑
5.30(m,1h),3.77
‑
3.68(m,1h),3.36
‑
3.16(m,3h),3.12
‑
3.03(m,1h),2.80
‑
2.63(m,6h),2.16
‑
2.02(m,1h),1.38
‑
1.28(m,1h),1.13
‑
0.72(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
9的结构式如上所示。
[0228]
实施例30
[0229]
本实施例提供一种化合物vii
‑
10,该化合物的结构式如下:
[0230]
[0231]
本实施例还提供一种化合物vii
‑
10的制备方法,包括以下步骤:
[0232]
中间体iv
‑
7的制备:本实施例提供的中间体iv
‑
7的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入2.27g、约10mmol的吲哚化合物iii
‑
7继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.93g黄色油状物iv
‑
7,产率为82%。其中,吲哚化合物iii
‑
7的结构式为中间体iv
‑
7的结构式为
[0233]
中间体v
‑
7的制备:本实施例提供的中间体iv
‑
7的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用4.79g、约10mmol的中间体iv
‑
7参加反应,得到4.1g白色固体v
‑
7,产率为91%。其中,中间体v
‑
7的结构式
[0234]
产物vii
‑
10的制备:将452mg中间体v
‑
7(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到51mg黄色油状物,产率为10%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
10;1h nmr(400mhz,dmso
‑
d6)δ12.05
‑
11.89(m,1h),10.84
‑
10.54(m,1h),10.25
‑
9.40(m,2h),8.68
‑
8.55(m,1h),7.76
‑
7.56(m,4h),7.49
‑
7.41(m,1h),7.39
‑
7.34(m,1h),7.30
‑
7.16(m,2h),7.15
‑
7.04(m,1h),6.94
‑
6.44(m,2h),5.70
‑
5.31(m,1h),3.79
‑
3.69(m,1h),3.40
‑
3.35(m,1h),3.28
‑
3.18(m,2h),3.11
‑
3.02(m,1h),2.79
‑
2.65(m,6h),2.23
‑
1.99(m,1h),1.48
‑
1.32(m,1h),1.20
‑
0.96(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
10的结构式如上所示。
[0235]
实施例31
[0236]
本实施例提供一种化合物vii
‑
11,该化合物的结构式如下:
[0237][0238]
本实施例还提供一种化合物vii
‑
11的制备方法,包括以下步骤:
[0239]
中间体iv
‑
8的制备:本实施例提供的中间体iv
‑
8的制备方法与实施例1提供的中
间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.31g、约10mmol的吲哚化合物iii
‑
8继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到2.61g黄色油状物iv
‑
8,产率为68%。其中,吲哚化合物iii
‑
8的结构式为中间体iv
‑
8的结构式为
[0240]
中间体v
‑
8的制备:本实施例提供的中间体iv
‑
8的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用3.84g、约10mmol的中间体iv
‑
8参加反应,得到3.52g白色固体v
‑
8,产率为99%。其中,中间体v
‑
8的结构式
[0241]
产物vii
‑
11的制备:将356mg中间体v
‑
8(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到41mg黄色油状物,产率为9%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
11;1h nmr(400mhz,dmso
‑
d6)δ11.46
‑
11.33(m,1h),10.29(s,1h),9.97
‑
9.61(m,1h),8.94
‑
8.83(m,1h),7.53
‑
7.34(m,2h),7.32
‑
7.26(m,1h),7.24
‑
7.11(m,2h),7.06
‑
6.94(m,1h),6.94
‑
6.50(m,2h),5.47
‑
5.27(m,1h),3.75
‑
3.62(m,1h),3.30
‑
3.20(m,2h),3.14
‑
3.05(m,1h),2.84
‑
2.66(m,7h),2.65
‑
2.54(m,1h),2.36
‑
2.16(m,3h),1.61
‑
1.48(m,1h),1.38
‑
1.21(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
11的结构式如上所示。
[0242]
实施例32
[0243]
本实施例提供一种化合物vii
‑
12,该化合物的结构式如下:
[0244][0245]
本实施例还提供一种化合物vii
‑
12的制备方法,包括以下步骤:
[0246]
中间体iv
‑
9的制备:本实施例提供的中间体iv
‑
9的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.35g、约10mmol的吲哚化合物iii
‑
9继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,
柱层析分离,得到2.68g黄色油状物iv
‑
9,产率为69%。其中,吲哚化合物iii
‑
9的结构式为中间体iv
‑
9的结构式为
[0247]
中间体v
‑
9的制备:本实施例提供的中间体iv
‑
9的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用3.88g、约10mmol的中间体iv
‑
9参加反应,得到3.6g白色固体v
‑
9,产率为100%。其中,中间体v
‑
9的结构式
[0248]
产物vii
‑
12的制备:将360mg中间体v
‑
9(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到58mg黄色油状物,产率为13%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
12;1h nmr(400mhz,dmso
‑
d6)δ11.75
‑
11.67(m,1h),10.60
‑
10.18(m,2h),10.08
‑
9.67(m,1h),9.17
‑
9.05(m,1h),7.67
‑
7.41(m,2h),7.40
‑
7.22(m,2h),7.22
‑
7.12(m,1h),7.03
‑
6.57(m,2h),5.63
‑
5.28(m,1h),3.74
‑
3.62(m,1h),3.34
‑
3.17(m,2h),3.16
‑
3.05(m,1h),2.86
‑
2.71(m,6h),2.70
‑
2.17(m,2h),1.65
‑
1.47(m,1h),1.34
‑
1.21(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
12的结构式如上所示。
[0249]
实施例33
[0250]
本实施例提供一种化合物vii
‑
13,该化合物的结构式如下:
[0251][0252]
本实施例还提供一种化合物vii
‑
13的制备方法,包括以下步骤:
[0253]
中间体iv
‑
10的制备:本实施例提供的中间体iv
‑
10的制备方法与实施例1提供的中间体iv
‑
1的制备方法基本相同,主要不同之处在于:反应体系中加入1.96g、约10mmol的吲哚化合物iii
‑
10继续常温搅拌6h。经tlc监测反应完全后,体系冷却至室温,加入硅胶炒样,柱层析分离,得到3.60g黄色油状物iv
‑
10,产率为80%。其中,吲哚化合物iii
‑
10的结构
式为中间体iv
‑
10的结构式为
[0254]
中间体v
‑
10的制备:本实施例提供的中间体iv
‑
10的制备方法与实施例1提供的中间体v
‑
1的制备方法基本相同,主要不同之处在于:采用4.49g、约10mmol的中间体iv
‑
10参加反应,得到4.13g白色固体v
‑
10,产率为98%。其中,中间体v
‑
10的结构式
[0255]
产物vii
‑
13的制备:将421mg中间体v
‑
10(约1mmol)和574mg hatu(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s1,同时将132mg n,n
‑
二甲基乙二胺(约1.5mmol)和195mg有机碱dipea(约1.5mmol)溶于dmf,常温搅拌1h,得到体系s2。将体系s2滴加至体系s1中,并继续常温反应6h。经tlc监测反应完全后,向体系中加入等量的水和20ml乙酸乙酯进行萃取,水相用乙酸乙酯萃取三次,合并有机层。有机层经无水硫酸镁干燥后,抽滤,加入硅胶炒样,柱层析分离,得到47mg黄色油状物,产率为10%。将该油状物溶于乙醇,加入2
‑
3滴浓盐酸,室温搅拌8h,减压蒸馏除去溶剂,即可得到盐酸盐产物vii
‑
13;1h nmr(400mhz,dmso
‑
d6)δ11.86
‑
11.74(m,1h),10.37(s,1h),10.21
‑
9.82(m,1h),9.09
‑
8.97(m,1h),7.97
‑
7.69(m,1h),7.59
‑
7.51(m,1h),7.43
‑
7.36(m,1h),7.34
‑
7.26(m,1h),7.25
‑
7.12(m,2h),7.04
‑
6.90(m,1h),6.80
‑
6.50(m,1h),5.53
‑
5.32(m,1h),3.75
‑
3.64(m,1h),3.39
‑
3.37(m,1h),3.32
‑
3.21(m,2h),3.14
‑
3.06(m,1h),2.81
‑
2.71(m,6h),2.68
‑
2.58(m,1h),1.65
‑
1.46(m,1h),1.37
‑
1.18(m,1h);因此,可以确定本实施例提供的所述化合物vii
‑
13的结构式如上所示。
[0256]
抑制lsd1活性的测定
[0257]
实验方法:样品为上述合成化合物vi
‑
1~vi
‑
20和vii
‑
1~vii
‑
13。样品储备液:分别称取样品1~2mg,用dmso(二甲基亚砜)溶解成浓度为10mm的母液,实验时用dmso稀释至需要测定浓度。分别将样品与从大肠杆菌表达体系中纯化获得的人源复合体蛋白lsd1/corest孵育,随后加入由吉尔生化(上海)有限公司合成的h3k4me2多肽孵育反应30min。孵育结束后,加入荧光染料amplex red和辣根过氧化物酶hrp反应5min,随后使用envision酶标仪(perkinelmer,waltham,ma,usa)测定荧光信号(e
x
=535nm,e
m
=595nm),并计算其抑制率。具体地,抑制率计算公式如下:
[0258][0259]
其中,上述抑制率计算公式中的“样品组荧光强度”、“标准组荧光强度”和“空白组荧光强度”均是采用参照上述实验方法测得的荧光强度数值,不同之处在于:在相同条件下,“样品组荧光强度”的测定对象是含待测上述化合物的样品液,“标准组荧光强度”的测
定对象是标准液,且该标准液与所述样品液相比不含有样品。“空白组荧光强度”的测定对象是不含lsd1/corest复合体蛋白和h3k4me2多肽的空白样品。用graphpad prism 8.0处理上述样品化合物的ic
50
数据,结果如表1所示。
[0260]
表1本发明实施例提供的化合物对lsd1/corest的抑制活性数据表
[0261][0262][0263]
最后应当说明的是:以上实施例仅用以说明本发明的技术方案而非对其限制;尽管参照较佳实施例对本发明进行了详细的说明,所属领域的普通技术人员应当理解:依然可以对本发明的具体实施方式进行修改或者对部分技术特征进行等同替换;而不脱离本发明技术方案的精神,其均应涵盖在本发明请求保护的技术方案范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1