一种罗库溴铵中间体的制备方法与流程
1.本发明属于药物合成技术领域,具体涉及一种罗库溴铵中间体的制备方法。
背景技术:
2.5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮是合成泮库溴铵、罗库溴铵、维库溴铵、哌库溴铵等非去极化甾体肌松药的重要中间体。罗库溴铵和维库溴铵等在内的甾醇溴铵类药物是近年来出现的一种新型的非去极化肌松药,作为麻醉辅助用药,用于气管内插管和手术中的肌肉松弛,具有起效快,持续时间短,无蓄积作用,不产生心动过速和血压变化,无组胺释放等特点。现有的常用制备5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮的方法大多采用表雄酮作为原料,通过磺酰化/消除两步法制备。
3.现有中国专利cn101684139公开了一种5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮的制备方法,该方法先制备中间产物表雄酮对甲苯磺酸酯,然后表雄酮对甲苯磺酸酯加入2,4,6
‑
三甲基吡啶回流反应脱去对甲苯磺酸酯,后处理以80%的较高收率得到5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮。2,4,6
‑
三甲基吡啶常压沸点171℃,在工业生产上蒸汽加热不容易达到回流温度,生产操作困难,无法工业化;2,4,6
‑
三甲基吡啶有刺激性,其急性毒性ld50为400mg/kg(大鼠经口),1000~2000mg/kg(豚鼠经皮),不宜于工业化应用;2,4,6
‑
三甲基吡啶价格昂贵,生产成本高。
4.现有中国专利cn 109678918a公开了5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮的制备方法,包括:以表雄酮(式1)为原料,在质子酸与三氟甲磺酸盐(三氟甲磺酸镧或三氟甲磺酸镱)共同催化下,在110℃下进行脱水反应,反应产物经后处理及重结晶分离得到深棕色油状的5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮(式2)。具体反应式如下:
[0005][0006]
该专利的制备方法是在传统工艺上进行了工艺叠缩,提高了生产效率,但是该工艺使用了昂贵的三氟甲磺酸镧或三氟甲磺酸镱这种三氟甲磺酸盐,同时使用甲苯在高温下长时间脱水反应得到的产品颜色深,需要经过重结晶的方式去除高温反应中引入的副产物。
[0007]
因此需要一种低温、高效、且成本较低的方法制备5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮。
技术实现要素:
[0008]
有鉴于此,本发明目的在于提供一种罗库溴铵中间体的制备方法,该中间体为5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮,该制备方法以表雄酮为原料,在伊顿试剂催化下进行脱水反应,该制备方法原料简单易得,该制备方法使用伊顿试剂(甲烷磺酸与五氧化二磷的混合物),将反应
温度降低至40℃,减少了长时间高温反应的副产物,即低双键异构杂质少,生产安全,三废少,适合工业化生产。
[0009]
所述制备方法包括:以式1所示的表雄酮为原料,在伊顿试剂催化下进行脱水反应生成所述罗库溴铵中间体,如式2所示;
[0010][0011]
优选地,所述表雄酮与所述伊顿试剂中的五氧化二磷摩尔比为1:0.1
‑
5。
[0012]
优选地,所述伊顿试剂中,五氧化二磷与甲烷磺酸的质量比为1:1
‑
10。
[0013]
优选地,所述伊顿试剂中,五氧化二磷与甲烷磺酸的质量比为1:9
‑
10。
[0014]
优选地,所述脱水反应的温度为30
‑
160℃,时间为6
‑
24h。
[0015]
优选地,所述脱水反应的温度为30
‑
40℃,时间为17
‑
24h。
[0016]
优选地,所述脱水反应中还可以加入溶剂,所述溶剂为二氯甲烷、甲苯、乙酸乙酯、丙酮、乙腈及二氧六环中的一种或多种。
[0017]
优选地,所述伊顿试剂中,五氧化二磷与甲烷磺酸的质量比为1:1
‑
2,更优选为1:1,所述溶剂为二氯甲烷、甲苯、乙酸乙酯、丙酮、乙腈及二氧六环中的一种或多种。
[0018]
进一步,所述脱水反应后经过洗涤、萃取、浓缩得到罗库溴铵中间体固体,即5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮固体。
[0019]
在某些具体实施例中,将脱水反应后的反应液倒入冰水中,加碳酸钠溶液洗涤,水相使用有机溶剂反萃取,合并有机相经无水硫酸钠干燥后,减压浓缩回收有机溶剂,得类白色固体。
[0020]
具体地,伊顿试剂的常规制备方法是甲烷磺酸和五氧化二磷重量比是1:10,使用五氧化二磷,甲烷磺酸这种强脱水性试剂,在一个较低的温度下可以通过延长反应时间将原料完全消耗。由于反应温度低,副产物少,水洗、萃取、干燥浓缩后即可得到类白色固体,高温反应得到的粗品必须要经过甲醇重结晶得到类白色固体,该操作会损失10%以上的收率。将甲烷磺酸的投料比降低至原先的10%,使用二氯甲烷做溶剂,该条件下原料能消耗完全,不经过重结晶即可得到类白色固体。
[0021]
本发明有益效果在于
[0022]
本发明提供的罗库溴铵中间体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮)的制备方法,通过使用伊顿试剂(甲烷磺酸与五氧化二磷的混合物),将反应温度降低至40℃,减少了长时间高温反应的副产物,反应液经过简单的水洗、萃取、浓缩即可得到类白色的固体,减少了重结晶去除副产物时造成的主产物损失。使得工艺更加简单、收率更低、三废排放少。
附图说明
[0023]
图1为实施例1制备的罗库溴铵中间体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮)纯度hplc图。
[0024]
图2为实施例2制备的罗库溴铵中间体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮)纯度hplc图。
[0025]
图3为实施例3制备的罗库溴铵中间体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮)纯度hplc图。
[0026]
图4为实施例3制备的罗库溴铵中间体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
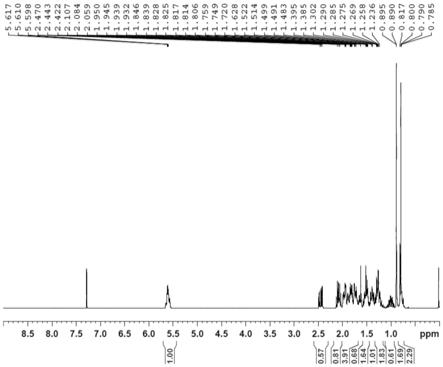
酮)的核磁图谱。
具体实施方式
[0027]
所举实施例是为了更好地对本发明进行说明,但并不是本发明的内容仅局限于所举实施例。所以熟悉本领域的技术人员根据上述发明内容对实施方案进行非本质的改进和调整,仍属于本发明的保护范围。
[0028]
本发明实施例中,纯度hplc的条件为:波长210nm;柱温:30℃;时间:35min;流速:1ml/min;流动相:甲醇:纯化水=85:15;色谱柱:triart c18 5um 250*4.6mm。
[0029]
实施例1
[0030]
将15.6g(0.11mol)五氧化二磷,甲烷磺酸(156g)加入洁净的250ml三口烧瓶中,室温搅拌2小时,然后加入表雄酮(29.0g,0.10mol),升温至110℃,反应6小时,tlc显示无原料。将反应液降低至室温,倒入500ml的水中,500ml二氯甲烷萃取,有机相使用250ml的5%碳酸钠水溶液洗涤两次,250ml纯化水洗涤有机相两次,无水硫酸钠干燥,过滤、减压浓缩回收二氯甲烷,得深棕色油状物,使用甲醇结晶得到21.6g类白色固体,hplc纯度大于95%,摩尔收率79.4%,其检测色谱结果如下表1所示,色谱图如图1所示。
[0031]
表1实施例1制备的5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮纯度hplc数据表
[0032][0033][0034]
实施例2
[0035]
将15.6g(0.11mol)五氧化二磷,甲烷磺酸(156g)加入洁净的250ml三口烧瓶中,室温搅拌2小时,然后加入表雄酮(29.0g,0.10mol),升温至40℃,反应17小时,tlc显示无原料。将反应液降低至室温,倒入500ml的水中,500ml二氯甲烷萃取,有机相使用250ml的5%碳酸钠水溶液洗涤两次,250ml纯化水洗涤有机相两次,无水硫酸钠干燥,过滤、减压浓缩回收二氯甲烷,得24.3g类白色固体,hplc纯度96.7%,摩尔收率89.3%,其检测色谱结果如下表2所示,色谱图如图2所示。
[0036]
表2实施例2制备的5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮纯度hplc数据表
[0037][0038]
实施例3
[0039]
将五氧化二磷(15.6g,0.11mol),甲烷磺酸(15.6g,0.16mol),二氯甲烷(300ml)加入洁净的500ml单口瓶中,室温搅拌2小时,然后加入表雄酮(29.0g,0.10mol),回流反应17小时。将反应液降低至室温,倒入500ml的水中,补加200ml二氯甲烷,有机相使用250ml的5%碳酸钠水溶液洗涤两次,250ml纯化水洗涤有机相两次,无水硫酸钠干燥,过滤、减压浓缩回收二氯甲烷,得24.6g类白色固体,摩尔收率90.4%,hplc纯度97.1%,水分小于0.1%。其检测色谱结果如下表3所示,色谱图如图3所示。
[0040]
表3实施例3制备的5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮纯度hplc数据表
[0041][0042]
实施例4
[0043]
将实施例3得到的类白色固体(5α
‑
雄甾
‑2‑
烯
‑
17
‑
酮)进行核磁共振分析,得到的核磁图谱如图4所示,所用的氘代试剂为cdcl3。
[0044]
最后说明的是,以上实施例仅用以说明本发明的技术方案而非限制,尽管参照较佳实施例对本发明进行了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的宗旨和范围,其均应涵盖在本发明的权利要求范围当中。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1