一种聚乙二醇单甲醚-聚乳酸嵌段共聚物及其制备方法与应用与流程
一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法与应用
技术领域
1.本发明涉及医药化工技术领域,具体地说,涉及一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法与应用。
背景技术:
2.聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物(mpeg
‑
pla)是一种两亲性的生物可降解材料,具有良好的生物相容性,无毒副作用,可生物降解,在疏水性药物的载体方面具有极高的应用价值。韩国samyang公司于2006年推出纳米胶束制剂该制剂即是使用聚合物mpeg
‑
pla包裹紫杉醇制备而成,于2002年由fda批准进入临床研究。该制剂体外和动物体内的生物相容性较好,胶束进入体内后迅速解离,释放药物,聚合物在体内可以在15小时内降解。平均分子量在3000
‑
5000的共聚物mpeg
‑
pla稳定性好,载药量高,适用于紫杉醇聚合物胶束的制备。
3.中国专利cn102219892公开了采用溶剂缩聚法(异丙醇、二甲苯、二甲基甲酰胺等溶剂)制备包含上述分子量的mpeg
‑
pla,但此类溶剂沸点较高,容易造成溶剂残留,影响进一步制备得到的药物聚合物胶束的安全性。目前不使用溶剂的熔融缩聚法是最常用的方法,如中国专利cn1214818c说明书制备例1所公开的方法,但得到的mpeg
‑
pla的分子量分布较宽,吸湿易结团,在与紫杉醇制备成紫杉醇聚合物胶束的过程中会产生较大的杂质,因此有必要进行制备方法的改进以提高产品质量。如中国专利cn104892909批露采用熔融缩聚法,其后采用纯水对二氯甲烷溶解的粗品进行水洗,有效除去水溶性低聚物,降低了产品分子量分布,并改善了吸湿性。
4.然而上述方法在聚合物的制备过程中,因加入催化剂而引入了大量的锡,由于该聚合物多作为药用辅料,对质量特别是重金属要求较高,为了使得产品质量可更符合药用标准,对于粗品的后处理效果仍有待提高。此外,上述方法中,产品制备环节均是在惰性气氛环境下的聚合,对起始物料的水分要求较高,反应体系除水较为麻烦,不利于工业化操作。
技术实现要素:
5.针对现有技术的不足,本发明的目的是提供一种可有效去除粗产品中的杂质、产率高,且制备过程简便、反应效果理想的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物的制备方法及应用。
6.为了实现该目的,本发明的技术方案如下:
7.一种制备聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物的方法,所述聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物由聚乙二醇单甲醚和丙交酯在辛酸亚锡催化下反应制得,反应结束后包括粗产品后处理步骤,所述粗产品后处理步骤包括:
8.(1)以第一析晶溶剂进行析晶,所述第一析晶溶剂为正己烷和无水乙醚的混合物;
9.(2)水洗;
10.(3)以第二析晶溶剂进行析晶,所述第二析晶溶剂为乙醚。
11.本发明中,聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物结构为:
[0012][0013]
其中,m为44,n为30
‑
36。
[0014]
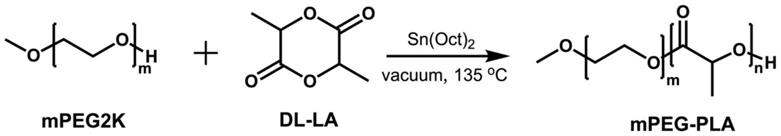
一个优选的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物的合成路线为:
[0015][0016]
其中,mpeg2k为平均分子量为2000的聚乙二醇单甲醚,本发明中还可使用分子量mn为1000~5000的mpeg。dl
‑
la为丙交酯。
[0017]
本发明经研究发现,在以辛酸亚锡作为催化剂的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物制备过程中,使用本发明的后处理方法,在保证高产率的前提下,可使产物中的丙交酯、乳酸和锡元杂质含量均大幅降低,提升了终产品的品质,使其更符合药用标准。
[0018]
本发明中,所述第一析晶溶剂中,正己烷和无水乙醚的体积比为(0.8
‑
1.5):1,优选为1:1,以配合后续步骤,更好地兼顾产率和除杂效果。
[0019]
本发明中,步骤(1)和(3)中的析晶在
‑
20℃至
‑
12℃下进行,优选为
‑
16℃,以获得更好的析晶除杂效果。
[0020]
本发明使用的第一析晶溶剂和第二析晶溶剂的温度为
‑
20℃至
‑
12℃,以保证析晶过程整体处于预期温度下。
[0021]
本发明中,步骤(1)和步骤(3)中析晶时,先以二氯甲烷溶解待析晶产物,然后在搅拌下将待析晶产物的二氯甲烷溶液加入到析晶溶剂中,所述搅拌的转速为200
‑
600rpm,以保证产率和除杂效果;步骤(2)中水洗时,也先以二氯甲烷溶解待水洗产物,再加入水进行洗涤;
[0022]
各析晶步骤中(步骤1和3),二氯甲烷的加入量以可完全溶解待析晶产物为宜,具体可为待析晶产物理论产量的0.5
‑
1.5倍体积当量,即每克待析晶产物,加入0.5
‑
1.5ml二氯甲烷。
[0023]
水洗步骤中(步骤2),二氯甲烷的加入量以可使待水洗产物充分溶解、分散为宜,具体可为待水洗产物理论产量的2
‑
2.5倍体积当量,即每克待水洗产物,加入2
‑
2.5ml二氯甲烷。
[0024]
优选,步骤(1)中,所述第一析晶溶剂的用量为步骤(1)中所用二氯甲烷体积的20倍;
[0025]
和/或,所述水洗时,所述水的用量为步骤(2)中所用二氯甲烷体积的0.8
‑
1.1倍,优选为1倍,以配合前后析晶步骤,保证除杂效果;
[0026]
和/或,所述第二析晶溶剂的用量为步骤(3)中所用二氯甲烷体积的15
‑
20倍。
[0027]
本发明中,所述聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物制备时,将聚乙二醇单甲醚、丙交酯和辛酸亚锡同时加入反应器中,先持续室温抽气保持体系真空,之后升温至50
‑
85℃,在搅拌下持续抽真空,再升温至110
‑
120℃,停止抽真空并维持搅拌,最后升温至聚合反应进行的温度。
[0028]
本发明中,所述聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物制备时,室温抽气的时间为2
‑
6h,在搅拌下抽真空的时间为20
‑
60min,停止抽真空维持搅拌的时间为15
‑
25min。
[0029]
本发明对聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物的制备环节进行了研究,通过特定的抽气步骤,使反应物可通过一锅法,在真空体系下熔融反应,制备得到分子量分布理想、产率高的目标产物。
[0030]
该方式对起始物料的水分要求较为宽松,水分含量不超过1%即可,反应起始物料勿需精制,可避免精制过程引入的溶剂,在一步加料后,可通过本发明的特定抽气流程,在充分、高效除原料中水分的同时,避免抽真空对原始物料总量的影响。既降低了体系水分对反应的影响,又因增加了反应体系的气密性,避免可能发生的气密性不好问题。
[0031]
作为一个优选制备方式,本发明将聚乙二醇单甲醚、丙交酯和辛酸亚锡同时加入反应器中,先持续室温抽气2h,之后升温至85℃,在搅拌下抽真空20min,再升温至120℃,停止抽真空维持搅拌20min,最后升温至聚合反应进行的温度。
[0032]
本发明中,聚乙二醇单甲醚和丙交酯的投料质量比为1:(1.15
‑
1.25),聚合反应的温度为130
‑
145℃,聚合反应的时间为24
‑
40h;
[0033]
优选,聚乙二醇单甲醚和丙交酯的投料质量比为1:1.2,聚合反应的温度为135℃,聚合反应的时间为40h,以获得理想的产率和聚合物产品。
[0034]
本发明还提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物,其由上述方法制备得到。
[0035]
本发明另提供一种上述聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物在作为疏水性药物载体中的应用。
[0036]
本发明的有益效果至少在于:
[0037]
本发明在保证高产率的情况下,可提高除去低聚物、催化剂及元素杂质的效果,使产品更加符合药用标准。且在制备反应时通过特定的抽气方式和真空熔融使聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物的制备既能通过一锅法进行,又无需对起始物料进行精制(对起始物料的水分要求较为宽松),既简化了制备操作流程,又能有效避免水对反应的影响,获得的产品经水洗析晶后,丙交酯、乳酸、锡含量明显降低,产品质量提升。
具体实施方式
[0038]
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
[0039]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0040]
实施例1
[0041]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0042]
具体包括如下步骤:
[0043]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌20min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0044]
粗产物精制过程:
[0045]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷(v为理论产量的体积当量,此处为每克待析晶产物,加入0.5ml二氯甲烷)溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的第一析晶溶剂中(第一析晶溶剂的体积为二氯甲烷体积的10倍,第一析晶溶剂由体积比为1:1的正己烷和无水乙醚组成),保持
‑
16℃沉淀,过滤收集白色固体粗品。
[0046]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中(每克上述白色固体粗品,加入2ml二氯甲烷),再加入以二氯甲烷的体积计1v的灭菌注射用水(即,加入水的体积为二氯甲烷体积的1倍),搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0047]
(3)在步骤(2)获得的粗品中加入理论产量0.5v的二氯甲烷(即,每克粗品,加入0.5ml二氯甲烷),之后将其在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的第二析晶溶剂——无水乙醚中,无水乙醚的用量为二氯甲烷体积的20倍,保持
‑
16℃析晶,过滤。
[0048]
(4)将步骤(3)的过滤产物30℃真空干燥,得终产物。
[0049]
实施例2
[0050]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0051]
具体包括如下步骤:
[0052]
聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物的制备方式同实施例1。精制过程如下:
[0053]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
12℃的第一析晶溶剂中(第一析晶溶剂的组成和加入量与实施例1相同),保持
‑
12℃沉淀,过滤收集白色固体粗品。
[0054]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中,再加入以二氯甲烷的体积计1v的灭菌注射用水,搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0055]
(3)以二氯甲烷(0.5v)溶解上述步骤(2)获得的产物(即,每克上述产物,加入0.5ml二氯甲烷),之后将溶解有上述产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
12℃的无水乙醚中(用量与实施例1相同),保持
‑
12℃沉淀,过滤收集白色固体产物。
[0056]
(4)将步骤(3)获得的产物30℃真空干燥,得终产物。
[0057]
实施例3
[0058]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0059]
具体包括如下步骤:
[0060]
聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物的制备方式同实施例1。精制过程如下:
[0061]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度200rpm)逐渐加至
‑
20℃的第一析晶溶剂中(第一析晶溶剂的组成和加入量与实施例1相同),保持
‑
20℃沉淀,过滤收集白色固体粗品。
[0062]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中,再加入以二氯甲烷的体积计1v的灭菌注射用水,搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0063]
(3)以二氯甲烷(0.5v)溶解上述步骤(2)获得的产物,之后将溶解有上述产物的二氯甲烷溶液在搅拌下(搅拌速度200rpm)逐渐加至
‑
20℃的无水乙醚中(用量与实施例1相同),保持
‑
20℃沉淀,过滤收集白色固体产物。
[0064]
(4)将步骤(3)获得的产物30℃真空干燥,得终产物。
[0065]
实施例4
[0066]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0067]
具体包括如下步骤:
[0068]
聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物的制备方式同实施例1。精制过程如下:
[0069]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的第一析晶溶剂中(第一析晶溶剂的体积为二氯甲烷体积的10倍,第一析晶溶剂由体积比为0.8:1的正己烷和无水乙醚组成),保持
‑
16℃沉淀,过滤收集白色固体粗产品。
[0070]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中,再加入以二氯甲烷的体积计1v的灭菌注射用水,搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0071]
(3)以二氯甲烷(0.5v)溶解上述步骤(2)获得的产物,之后将溶解有上述产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的无水乙醚中(用量与实施例1相同),保持
‑
16℃沉淀,过滤收集白色固体产物。
[0072]
(4)将步骤(3)获得的产物30℃真空干燥,得终产物。
[0073]
实施例5
[0074]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0075]
具体包括如下步骤:
[0076]
聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物的制备方式同实施例1。精制过程如下:
[0077]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的第一析晶溶剂中(第一析晶溶剂的体积为二氯甲烷体积的10倍,第一析晶溶剂由体积比为1.5:1的正己烷和无水乙醚组成),保持
‑
16℃沉淀,过滤收集白色固体粗品。
[0078]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中,再加入以二氯甲烷的体积计1v的灭菌注射用水,搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0079]
(3)以二氯甲烷(0.5v)溶解上述步骤(2)获得的产物,之后将溶解有上述产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的无水乙醚中(用量与实施例1相同),保持
‑
16℃沉淀,过滤收集白色固体产物。
[0080]
(4)将步骤(3)获得的产物30℃真空干燥,得终产物。
[0081]
实施例6
[0082]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0083]
具体包括如下步骤:
[0084]
聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物的制备方式同实施例1。精制过程如下:
[0085]
(1)反应器中加入待析晶产物(粗产物)理论产量0.5v的二氯甲烷溶解产物,之后将溶解有产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃第一析晶溶剂中(第一析晶溶剂的组成和加入量与实施例1相同),保持
‑
16℃沉淀,过滤收集白色固体粗品。
[0086]
(2)将上述白色固体粗品溶于二氯甲烷(2v)中,再加入以二氯甲烷的体积计0.8v的灭菌注射用水,搅拌40min,分层,分出有机相,旋转蒸发溶剂。
[0087]
(3)以二氯甲烷(0.5v)溶解上述步骤(2)获得的产物,之后将溶解有上述产物的二氯甲烷溶液在搅拌下(搅拌速度600rpm)逐渐加至
‑
16℃的无水乙醚中(用量与实施例1相同),保持
‑
16℃沉淀,过滤收集白色固体产物。
[0088]
(4)将步骤(3)获得的产物30℃真空干燥,得终产物。
[0089]
实施例7
[0090]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0091]
具体包括如下步骤:
[0092]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至50℃,开启搅拌(搅拌速率为100rpm)并抽真空60min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌15min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0093]
精制过程与实施例1相同。
[0094]
实施例8
[0095]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0096]
具体包括如下步骤:
[0097]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌25min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0098]
精制过程与实施例1相同。
[0099]
实施例9
[0100]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0101]
具体包括如下步骤:
[0102]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌
20min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应24h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0103]
精制过程与实施例1相同。
[0104]
实施例10
[0105]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0106]
具体包括如下步骤:
[0107]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌20min(搅拌速率为500rpm)。最后,升温至130℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0108]
精制过程与实施例1相同。
[0109]
实施例11
[0110]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0111]
具体包括如下步骤:
[0112]
将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌20min(搅拌速率为500rpm)。最后,升温至145℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0113]
精制过程与实施例1相同。
[0114]
实施例12
[0115]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0116]
具体包括如下步骤:
[0117]
将400g mpeg(平均分子量2000)、460g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌20min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0118]
精制过程与实施例1相同。
[0119]
实施例13
[0120]
本实施例提供一种本发明的聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法。
[0121]
具体包括如下步骤:
[0122]
将400g mpeg(平均分子量2000)、500g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。室温下抽气2h使体系保持真空。升温至85℃,开启搅拌(搅拌速率为100rpm)并抽真空20min。之后,升温至120℃并停止抽真空,维持体系真空状态下继续搅拌20min(搅拌速率为500rpm)。最后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0123]
精制过程与实施例1相同。
[0124]
对比例1
[0125]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:在步骤(1)中选用的第一析晶溶剂为正己烷。观察发现,第一次析晶后产物粘度大,抽滤时随溶剂滤出。
[0126]
对比例2
[0127]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:在步骤(1)中选用的第一析晶溶剂为乙醚。
[0128]
对比例3
[0129]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:在步骤(1)中,第一析晶溶剂由体积比为1:1的乙醇和无水乙醚组成。
[0130]
对比例4
[0131]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:在步骤(1)中,第一析晶溶剂由体积比为2:1的正己烷和无水乙醚组成。观察发现,第一次析晶后产物略粘稠。
[0132]
对比例5
[0133]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:步骤(2)中水的用量以二氯甲烷的体积计为2倍体积当量。
[0134]
对比例6
[0135]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:重复进行步骤(2),即共进行2次相同的水洗步骤。
[0136]
对比例7
[0137]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:步骤(1)和步骤(3)中的析晶温度为0℃。
[0138]
对比例8
[0139]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:步骤(2)中水的用量以二氯甲烷的体积计为0.5倍体积当量。
[0140]
对比例9
[0141]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:不进行步骤(2),仅通过两次析晶来进行精制。
[0142]
对比例10
[0143]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,精制时:步骤(1)和步骤(3)中的析晶搅拌速度为100rpm。
[0144]
对比例11
[0145]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,制备粗产品时,将400g mpeg(平均分子量2000)、480g丙交酯(dl
‑
la)及2.0g辛酸亚锡(sn(oct)2)一同加入到反应器中。在100℃下抽真空除水40min。
之后,升温至135℃,聚合反应40h。反应结束后,冷却反应体系获得聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物粗产物。
[0146]
精制过程与实施例1相同。
[0147]
对比例12
[0148]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,制备粗产品时,聚合反应时间为15h。
[0149]
对比例13
[0150]
本对比例提供一种聚乙二醇单甲醚
‑
聚乳酸嵌段共聚物及其制备方法,其制备方法与实施例1相同,区别仅在于,先将mpeg与辛酸亚锡加入反应瓶,升至120℃抽真空20min,然后再加入dl
‑
la,再抽真空20min。之后进一步升温进行聚合反应。
[0151]
实验例1
[0152]
本实验例对各实施例和对比例制得的成品进行检测。
[0153]
测试方法如下:
[0154]
产率的计算方法为:实产量/理论产量。
[0155]
gpc(pd)的测试方法按照分子排阻色谱法(通则0541)测定,具体如下:
[0156]
供试品溶液:取本品适量,精密称定,加四氢呋喃溶解制成每1ml中约含2mg的溶液,混匀,滤过,作为供试品溶液。
[0157]
对照品溶液:取聚苯乙烯分子量对照品(分子量1920、2790、4750、6660、12980)适量,加四氢呋喃溶解并制成每1ml中约含2mg的溶液,摇匀,作为对照品溶液。
[0158]
色谱条件:用凝胶色谱柱(waters styragelguard ht 3 thf 4.0*6.0mm、waters styragelht 3 thf 7.8*300mm,500a和waters styragelht 5 dmf 7.8*300mm,500a串联),以四氢呋喃为流动相,示差折光检测器;检测器温度为35℃,柱温为30℃,进样体积为100μl,流速1.0ml/min,示差折光检测器。
[0159]
测定法:取上述对照品溶液,注入液相色谱仪,记录色谱图,由gpc软件计算回归方程,线性相关系数r应不得小于0.99。取供试品溶液,同法测定,根据回归方程计算供试品的重均分子量及分子量分布。本发明具体实施方式部分所用的聚乙二醇单甲醚的分子量为2000;聚合反应后,以上述gpc方法测得的mn值在5500
±
500为适宜的数值。
[0160]
丙交酯照高液相色谱法(通则0512)测定,具体如下:
[0161]
对照品溶液:取丙交酯约25mg,精密称定,置50ml量瓶中,加乙腈溶解并稀释至刻度,摇匀;精密量取3ml,置10ml量瓶中,加乙腈稀释至刻度,摇匀,滤过,作为对照品溶液。
[0162]
供试品溶液:取本品约100mg,精密称定,置10ml量瓶中,加乙腈溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
[0163]
色谱条件:以十八烷基硅烷键合硅胶为填充剂(如waters xbridge c18 4.6mm
×
150mm,5μm或其他效能相当的色谱柱),以乙腈
‑
水(10:90)为流动相,流速为1.0ml/min;柱温为35℃;检测波长为210nm,进样体积为10μl。
[0164]
测定法:取对照品溶液和供试品溶液,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算,即得。
[0165]
乳酸照高液相色谱法(通则0512)测定,具体如下:
[0166]
对照品溶液:取乳酸约50mg,精密称定,置50ml量瓶中,加水溶解并稀释至刻度,摇
匀;精密量取1ml,置100ml量瓶中,加水稀释至刻度,摇匀,滤过,作为对照品溶液。
[0167]
供试品溶液:取本品约20mg,精密称定,置10ml量瓶中,加水溶解并稀释至刻度,摇匀,滤过,作为供试品溶液。
[0168]
色谱条件:以十八烷基硅烷键合硅胶为填充剂(如welch ultimate aq
‑
c18 4.6mm*250mm,5μm或其他效能相当的色谱柱),以乙腈
‑
0.12%磷酸水溶液(2:98)为流动相,流速为1.0ml/min;柱温为35℃;检测波长为210nm,进样体积为20μl。
[0169]
测定法:取上述对照品溶液和供试品溶液,分别注入液相色谱仪,记录色谱图,按外标法以峰面积计算,即得。
[0170]
锡的测试方法:icp
‑
ms法。
[0171]
仪器:icp
‑
ms agilent 7900。
[0172]
元素混合标准母液:分别移取li、ti、v、cr、mn、co、ni、cu、as、sn、nb、mo、pd、cd、sb、ba、pt、tl、pb元素标准溶液(1mg/ml)0.1ml置于100ml量瓶中,加5%稀硝酸稀释,定容至刻度,摇匀,备用。
[0173]
工作标准曲线溶液:分别精密移取元素混合标准母液(1mg/l)适量,置于25ml量瓶中,5%稀硝酸定容,配制成系列浓度的不同浓度的标准溶液(0.1μg/l,5μg/l、10μg/l、30μg/l、50μg/l)。
[0174]
供试品溶液:取本品约0.5g,加浓硝酸10ml,于通风橱内,置电热板上160℃加热30min,待温度冷却至室温,将上述溶液转移至50ml容量瓶中,5%稀硝酸稀释定容,即得。结果如下表1:
[0175]
表1
[0176]
[0177][0178]
从表1中可知,对比例1、3、5、6、7、12产率大幅降低,损失大。
[0179]
对比例2终产物状态较好,但其锡元杂质、丙交酯和乳酸含量高,超过药用限度。
[0180]
对比例4一次析晶后产物略粘稠,对最终产率有较大影响。
[0181]
对比例7产物状态较差,粘度大,不是良好的固体,抽滤时损失大。
[0182]
对比例8产物锡元素杂质、丙交酯与乳酸含量高。
[0183]
对比例9未用水洗,产物的分子量分布宽,pd值高;锡元素杂质含量过高,不满足药用需求。
[0184]
对比例10产物状态粘度大,具一定流动性,抽滤损失大。
[0185]
对比例11反应前处理方式不合适,造成产物分子量低且产率低。
[0186]
对比例12中聚合反应未完全,分子量低且产率低。
[0187]
对比例13的条件下,产物产率低且相对分子量低。
[0188]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1