基于宏基因组学快速确定微生物的方法及系统与流程
1.本发明涉及病原检测技术领域,尤其涉及一种基于宏基因组学(mngs)快速确定微生物的方法及系统。
背景技术:
2.呼吸道感染是全球范围内临床最常见的疾病之一,根据其部位分为上呼吸道感染和下呼吸道感染。前者包括鼻炎、咽炎和喉炎;后者包括气管炎、支气管炎和肺炎。根据全球疾病死亡率调查,急性呼吸道感染造成的死亡人数位居所有疾病造成死亡数的前列。引起呼吸道感染的病原体种类很多,其中包括细菌、真菌、病毒、支原体、衣原体等,其临床表现与治疗措施存在差异。最常见的呼吸道病原体包括甲型流感病毒、乙型流感病毒、副流感病毒、呼吸道合孢病毒、流感嗜血杆菌、肺炎军团菌、肺炎链球菌、结核分枝杆菌、念珠菌、霉菌、肺炎支原体、肺炎衣原体等。最近几年,又有新的呼吸道病原体在不断地被发现,如高致病性禽流感病毒、甲型流感毒h1n1以及新型冠状病毒2019-ncov等。
3.呼吸道病原体的检测方法很多,为临床疾病诊断和治疗提供了一定帮助,目前常见的临床检测方法主要有:病毒培养分离法、酶联免疫吸附法(elisa)、聚合酶链反应(pcr)等。这些方法只能针对已知特殊的病原体检测,检测时间较长、成本较高,其阳性率不高或存在家阳性率情况。而mngs(宏基因组测序)技术在发现和转化研究的诸多方面已逐渐成为一种被广泛采用的技术,在急难危重感染性疾病的病原诊断分析中有重要应用,能够对样本中的所有核酸进行无偏向测序,包括人源和微生物的核酸。可以快速高效的检测样品中病原体种类。
4.然而通过宏基因组测序在呼吸道疾病的快速检测还需要进一步改进。
技术实现要素:
5.本发明旨在至少在一定程度上解决相关技术中的技术问题之一。为此,本发明提供了一种基于宏基因组学快速确定微生物的方法和系统。通过本发明所提供的确定微生物的方法和系统可以高效、快速进行呼吸道感染的检测。
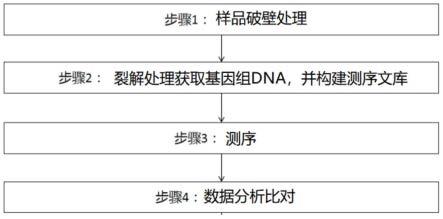
6.本发明的第一方面提供了一种基于宏基因组学快速确定微生物的方法,所述方法包括:
7.(1)对待测样品进行破壁处理,以便获得破壁后产物,所述破壁处理包括酶解处理和物理破壁处理;
8.(2)对所述破壁后产物进行裂解处理,获取来自于所述待测样品的基因组dna,并基于所述基因组dna构建测序文库;
9.(3)对所述测序文库进行测序,以便获得测序数据;
10.(4)基于所述测序数据,与微生物数据库比对,确定所述待测样品中的微生物信息。
11.根据本发明的实施例,以上所述基于宏基因组学快速确定微生物的方法还可以进
一步包括如下技术特征:
12.根据本发明的实施例,所述酶解处理包括通过溶菌酶和溶壁酶处理。
13.根据本发明的实施例,所述溶菌酶的添加量为500-3000u;所述溶壁酶的添加量为20-200u。所提到的u为酶活蛋白。根据本发明的实施例,溶菌酶的添加量可以为500u~2500u、1000u~2500u、1500u~2500u、2000u~2500u等。溶壁酶的添加量为40~200u、60~200u、80~200u、100~200u、120~200u、140~200u、160~200u、180~200u等。
14.根据本发明的实施例,所述酶解处理的条件为37摄氏度条件下酶解10~30分钟。
15.根据本发明的实施例,所述物理破壁处理包括通过将待测样品和玻璃珠混合,进行高速震荡的步骤。
16.根据本发明的实施例,所述高速震荡的条件为50~80hz的速度震荡1~3分钟,时间间隔为10~60秒,1~10个循环。
17.根据本发明的实施例,通过下述步骤获取所述待测样品的基因组dna:将所述破壁后产物和载体rna(carrier rna)、裂解酶k和缓冲液混合,65℃下进行裂解反应,以便获得反应液;将所述反应液进行纯化,去除盐离子和有机试剂,以便获得来自于所述待测样品的基因组dna。通过和载体rna混合,可以提高微量样品中dna的得率。
18.根据本发明的实施例,基于所述基因组dna通过下述步骤构建测序文库:将所述基因组dna进行酶切片段化处理,获得酶切产物;将所述酶切产物和测序接头连接,获得连接产物;对所述连接产物进行纯化与质控,以便得到测序文库。
19.根据本发明的实施例,所述酶切片段化处理包括采用非限制性核酸内切酶进行处理。
20.根据本发明的实施例,所述酶切片段化处理的反应体系为:50重量份基因组dna和10重量份非限制性核酸内切酶混合。
21.根据本发明的实施例,所述酶切片段化处理的条件为在30
±
1℃的条件下孵育15
±
5min进行片段化,然后在72
±
1℃的条件下孵育25
±
5min进行变性处理;
22.根据本发明的实施例,所述基因组dna的用量为50-200ng。
23.根据本发明的实施例,所述连接反应体系为:60重量份所述酶切产物、30重量份连接缓冲液、5重量份核酸连接酶和5重量份测序接头核酸分子混合。
24.根据本发明的实施例,所述连接反应条件为在20
±
1℃的条件下孵育25
±
5min。
25.根据本发明的实施例,所述测序接头为退火y型全长unique dual index(udi),所述测序接头双端含8nt唯一样本标签(index)。
26.根据本发明的实施例,步骤(3)中所述测序处理包括:测序文库定量,并稀释为0.5-3.5pm,在测序仪上进行高通量测序的步骤。
27.根据本发明的实施例,步骤(4)进一步包括:(4-1)基于特异性样本标签序列,将所述测序数据进行拆分,所述特异性样本标签序列用于区分不同的样本;(4-2)将测序数据和宿主数据库比对,计算测序数据中的宿主序列比例,以便去除宿主序列;(4-3)将去除宿主序列后的序列与微生物数据库进行比对,确定待测样品中的微生物信息。当对多个样品进行建库测序时,利用特异性标本标签序列区分不同的样品,可以同时实现对多个样品同时进行测序。
28.根据本发明的实施例,所述待测样品选自痰液、肺泡灌洗液、鼻腔粘液类样本中的
至少一种。
29.本发明的第二方面提供了一种基于宏基因组学快速确定微生物的系统,所述系统包括:
30.预处理模块,所述预处理模块用于对待测样品进行破壁处理,以便获得破壁后产物,所述破壁处理包括酶解处理和物理破壁处理;
31.测序文库构建模块,所述测序文库构建模块用于对所述破壁后产物进行裂解处理,获取来自于所述待测样品的基因组dna,并基于所述基因组dna构建测序文库;
32.测序模块,所述测序模块用于对所述测序文库进行测序,以便获得测序数据;
33.数据分析模块,所述数据分析模块基于所述测序数据,与微生物数据库比对,确定所述待测样品中的微生物信息。
34.根据本发明的实施例,以上所述的基于宏基因组学快速确定微生物的系统可以进一步包括如下技术特征:
35.根据本发明的实施例,以上基于宏基因学快速确定微生物的系统中,所述酶解处理包括通过溶菌酶和溶壁酶处理;
36.根据本发明的实施例,以上基于宏基因学快速确定微生物的系统中,所述溶菌酶的添加量为500-3000u;所述溶壁酶的添加量为20-200u。
37.根据本发明的实施例,以上基于宏基因学快速确定微生物的系统中,所述酶解处理的条件为37摄氏度10~30分钟。
38.根据本发明的实施例,以上基于宏基因学快速确定微生物的系统中,所述物理破壁处理包括通过将待测样品和玻璃珠混合,进行高速震荡的步骤。
39.根据本发明的实施例,以上基于宏基因学快速确定微生物的系统中,所述高速震荡的条件为50~80hz的速度震荡1~3分钟,时间间隔为10~60秒,1~10个循环。
40.根据本发明的实施例,所述测序文库构建模块通过下述步骤获取所述预处理样品中的核酸:
41.将所述破壁后产物和carrier rna、裂解酶k和缓冲液混合,65℃下进行裂解反应,以便获得反应液;
42.将所述反应液进行纯化,去除盐离子和有机试剂,以便获得所述待测样品的基因组dna。
43.根据本发明的实施例,所述测序文库构建模块通过下述单元获得测序文库:片段化处理单元,所述片段化处理单元用于将所述基因组dna进行酶切片段化处理,获得酶切产物;连接单元,所述连接单元用于将所述酶切产物和测序接头连接,获得连接产物;纯化和质控单元,所述纯化和质控单元用于对所述连接产物进行纯化与质控,以便得到测序文库。
44.根据本发明的实施例,所述酶切片段化处理包括采用非限制性核酸内切酶进行处理。
45.根据本发明的实施例,所述酶切片段化处理的反应体系为:50重量份基因组dna和10重量份非限制性核酸内切酶混合。
46.根据本发明的实施例,所述酶切片段化处理的条件为在30
±
1℃的条件下孵育15
±
5min进行片段化,然后在72
±
1℃的条件下孵育25
±
5min进行变性处理。
47.根据本发明的实施例,所述基因组dna的用量为50-200ng。
48.根据本发明的实施例,所述连接反应体系为:60重量份所述酶切产物、30重量份连接缓冲液、5重量份核酸连接酶和5重量份测序接头混合。根据本发明的实施例,所述连接反应条件为在20
±
1℃的条件下孵育25
±
5min。
49.根据本发明的实施例,所述测序接头为退火y型全长unique dual index(udi),所述测序接头双端含8nt唯一样本标签(index)。
50.根据本发明的实施例,所述测序模块包括:将定量稀释为0.5-3.5pm的测序文库在测序仪上进行高通量测序的步骤。
51.根据本发明的实施例,所述数据分析模块进一步包括:数据拆分单元,所述数据拆分单元基于特异性样本标签序列,将所述测序数据进行拆分,所述特异性样本标签序列用于对不同样本进行区分;宿主比对单元,所述宿主比对单元用于将测序数据和宿主数据库比对,计算测序数据中的宿主序列比例,去除宿主序列;微生物比对单元,所述微生物比对单元用于将去除宿主序列后的序列与病原微生物数据库进行比对,确定待测样品中的微生物信息。
52.本发明具有以下有益效果:
53.(1)本发明所提供的方法或者系统,不需要对呼吸道感染病原微生物样品进行培养,只需对样品进行提取处理后,对所提取的dna样品进行测序,获取核酸序列信息,根据核酸序列信息便可获取病原微生物样品的检测结果,大大缩短了检测所耗费的时间,从而实现了对病原微生物样品的快速检测。
54.(2)本发明所提供的样品破壁方法,通过使用酶解方法与物理破壁方法结合对微生物细胞进行细胞壁破碎处理,能够高效的破碎真菌、结核类杆菌、革兰氏阳性菌的细胞壁,提高这类病原体的检测灵敏度。
55.(3)本发明所提供的基于mngs的高通量测序文库构建方法,使用片段化酶对dna进行片段化,dna的损失较机械法更小,同时片段化的速率也更快,另外采用pcr-free方法构建,避免pcr扩增带来非特异性扩增偏好性,提高了整个文库的构建效率和检测灵敏度。
56.(4)本发明所提供的基于mngs的病原微生物检测方法和系统,通过与病原微生物数据库的比对,可以得到更准确的病原微生物匹配结果。
57.本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
58.本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
59.图1为本发明所提供的基于宏基因组学快速确定微生物的方法的步骤图。
60.图2为根据本发明的实施例提供的基于宏基因组学快速确定微生物的系统的结构示意图。
61.图3为根据本发明的实施例提供的测序文库构建模块的结构示意图。
具体实施方式
62.下面详细描述本发明的实施例,所述实施例的示例在附图中示出。下面通过参考
附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
63.本发明提供了一种基于宏基因组学快速确定微生物的方法,所述方法包括:(1)对待测样品进行破壁处理,以便获得破壁后产物,所述破壁处理包括酶解处理和物理破壁处理;(2)对所述破壁后产物进行裂解处理,获取来自于所述待测样品的基因组dna,并基于所述基因组dna构建测序文库;(3)对所述测序文库进行测序,以便获得测序数据;(4)基于所述测序数据,与微生物数据库比对,确定所述待测样品中的微生物信息。
64.在一些实施方式中,步骤(1)中采用酶解处理与物理破壁处理联合对待测样品进行破壁处理。酶解处理和物理破壁处理的顺序不做特殊要求。根据本发明的实施例,先对待测样品进行酶解处理,然后进行物理破壁处理。在一些具体实施方式中,待测样品中加入500-3000u溶菌酶和20-200u溶壁酶,混匀,37℃温育10-30min。酶解处理之后采用物理破壁的方式进行处理。在一些实施方式中,向经过酶解处理的样品中加入玻璃珠,使用均质仪高速震荡处理。高速震荡处理的参数可以根据实际需要进行调整。在一些优选实施方式中,采用均质仪进行高速震荡处理的条件可以设置为:50-80hz的速度振荡1-3min,间隔10-60s,共1-10个循环。通过酶解方法和物理破壁处理的方法破碎微生物细胞壁,可提高破壁效率,确保后续提取、建库效果。
65.待测样品的类型不受特别限制,可以为痰液、肺泡灌洗液、鼻腔粘液等可以用来检测呼吸道感染的样品。
66.在一些实施方式中,通过下述步骤获取所述待测样品的基因组dna:将所述破壁后产物和carrier rna、裂解酶k(lysis enzyme k)和缓冲液混合,在65℃下进行裂解反应,以便获得反应液;将所述反应液进行纯化,去除盐离子和有机溶剂,以便获得基因组dna。通过裂解酶k对核酸对破壁后的样品进行裂解处理。然后进行纯化,可以进一步提高核酸的质量,确保后续建库的效果。在一些具体实施方式中,所述裂解反应在65摄氏度条件下反应15分钟,然后再在4摄氏度条件下反应5分钟。
67.可以通过本领域常用的方法对反应液进行纯化。例如可以通过一些商用的试剂盒进行纯化。纯化后的产物可以进行干燥处理、洗脱。这些均可以通过本领域常用的方法或者一些商用的试剂盒即可以完成。
68.基于所述基因组dna通过下述步骤构建测序文库:将所述基因组dna进行酶切片段化处理,获得酶切产物;将所述酶切产物和测序接头连接,获得连接产物;对所述连接产物进行纯化与质控,以便得到测序文库。
69.在一些实施方式中,可以将50重量份基因组dna样品和10重量份非限制性核酸内切酶混合,进行酶切片段化处理。例如可以在30
±
1℃的条件下孵育15
±
5min进行片段化,然后在72
±
1℃的条件下孵育25
±
5min进行变性处理。酶切片段化处理时所用到的基因dna的总量可以在50-200ng范围内;例如基因组dna的样品总量为100ng。酶切片段化处理中非限制性核酸内切酶mix为dna smearase mix。
70.所述连接反应体系为:60重量份酶切后产物、30重量份连接缓冲液、5重量份核酸连接酶和5重量份测序接头混合进行连接反应。在一些实施方式中,所述连接反应条件为在20
±
1℃的条件下孵育25
±
5min。所用到的测序接头可以为退火y型全长unique dual index(udi),其双端含8nt唯一样本标签(index)。所用到的核酸连接酶本领域常用的连接酶,例如可以为t4 dna连接酶。连接后的产物可以通过本领域常用的方法进行纯化,例如可
以通过磁珠进行纯化。如以0.6
×
peg浓度缓冲液羧基磁珠进行纯化,用30-40ul蒸馏水进行洗脱。通过该方法所获得的测序文库片段主峰位于300-500bp处,测定结果更加精确。
71.在对测序文库进行测序之前,可以对测序文库进行定量、混合,包括以下步骤:对纯化后所得测序文库进行片段大小和浓度检测;根据测序文库的片段大小和浓度检测结果进行摩尔浓度计算;再根据测序文库所需测得的数据量大小,进行混合体积计算;按照计算得到的体积进行混合实验,即得到上机测序文库。在一些实施方式中,纯化后所得测序文库的片段大小检测采用agilent 2100bioanalyzer仪器检测。在一些实施方式中,对纯化后所得测序文库的浓度检测采用qubit 4.0仪器检测。混合后的测序文库的个数大约在20个左右。
72.其中,步骤(3)中所述测序处理包括:将定量稀释为0.5-3.5pm的测序文库在测序仪上进行高通量测序的步骤。所述高通量测序包括边合成边测序或边连接边测序。所用到的测序仪为illumina nextseq dx。
73.在一些实施方式中,步骤(4)进一步包括:(4-1)基于特异性index序列,对所述测序数据进行拆分;(4-2)将测序数据和宿主数据库比对,计算测序数据中的宿主序列比例,去除宿主序列;(4-3)将去除宿主序列后的序列与微生物数据库进行比对,确定待测样品中的微生物信息。所提到的拆分方法可以为,每一个测序文库两端所连接的接头上都有一个特异的双index序列,数据下机后,会根据每一条序列上测得的双index序列将不同的reads分配至不同的测序文库下。在将测序数据和宿主数据库比对时,首先构建人源数据库;将每个测序文库的reads序列信息与宿主数据库比对,计算数据中的宿主序列比例,然后将比对到的宿主序列剔除,得到不含宿主序列的reads。所提到的宿主数据库优选为人源数据库。将去除宿主序列后的序列与微生物数据库进行比对为,首先构建微生物数据库;将不含宿主序列的reads序列与微生物数据库比对,以确定所述微生物样品中的微生物种类及含量。
74.通过所提供的方法不仅可以用来检测待测样品中的微生物种类,不仅可以用来确定病原微生物的种类,还可以用来确定非导致疾病的微生物的种类。所提供的基于宏基因组学快速确定微生物的方法可以用于疾病的诊断和治疗,也可以用于非疾病的诊断和治疗。
75.本发明还提供了一种基于宏基因组学快速确定微生物的系统,如图2所示,所述系统包括:破壁处理模块,所述破壁处理模块用于对待测样品进行破壁处理,以便获得破壁后产物,所述破壁处理包括酶解处理和物理破壁处理;测序文库构建模块,所述测序文库构建模块和所述破壁处理模块连接,所述测序文库构建模块用于对所述破壁后产物进行裂解处理,获取来自于所述待测样品的基因组dna,并基于所述基因组dna构建测序文库;测序模块,所述测序模块和所述测序文库构建模块相连,所述测序模块用于对所述测序文库进行测序,以便获得测序数据;数据分析模块,所述数据分析模块和所述测序模块相连,所述数据分析模块基于所述测序数据,与微生物数据库比对,确定所述待测样品中的微生物信息。
76.在一些实施方式中,所述测序文库构建模块通过下述单元获得测序文库,如图3所示:片段化处理单元,所述片段化处理单元用于将所述基因组dna进行酶切片段化处理,获得酶切产物;连接单元,所述连接单元和所述片段化处理单元相连,所述连接单元用于将所述酶切产物和测序接头连接,获得连接产物;纯化和质控单元,所述纯化和质控单元和所述连接单元相连,所述纯化和质控单元用于对所述连接产物进行纯化与质控,以便得到测序
文库。
77.以下将通过实施例对本发明进行详细描述。需要说明的是,这些实施例仅用于方便本领域技术人员理解,不应看做是对本发明保护范围的限制。实施例中未提及的具体的参数或者步骤均可以通过本领域的常识获得,而且所用到的试剂均可以通过商用获得。
78.实施例1
79.实施例1以自制阳性企业参考品作为实验样本,参考品含约1
×
10
10
cfu总量的微生物细胞混合物,尝试利用不同的方法来确定样本中的微生物。其中微生物细胞混合物包含:铜绿色假单胞菌、大肠肝菌、鼠伤寒沙门菌、乳杆菌、粪肠球菌、金黃葡萄球菌、李斯特氏菌、枯草芽孢杆菌、酿酒酵母、隐球菌。其中各个微生物细胞含量见下表1所示。
80.表1微生物细胞量
[0081][0082][0083]
制备例1
[0084]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0085]
(1)样品前处理i:取样品0.8ml,加入10μl溶菌酶(10mg/ml)和10μl溶壁酶(10000u/ml),混匀,37℃温育20min;
[0086]
(2)样品前处理ii:再加入200mg玻璃珠,使用均质仪高速震荡处理,其均质仪参数可设置为:60hz的速度振荡1min,间隔30s,共5个循环;
[0087]
(3)8000rpm离心30s,转移上清600ul置新2ml离心管中;
[0088]
(4)向2ml离心管中加入2ul carrier rna(1ug/ul)储存液,60ul蛋白酶k和600ul缓冲液gb,震荡混合15s,低速瞬离5s以去除管盖内壁的液滴;
[0089]
(5)在65℃温育15min,并不时摇动样品。低速瞬离5s以去除管盖内壁的液滴;
[0090]
(6)待样品温度降至室温后加入600μl预冷的无水乙醇,并涡旋混匀15s,室温放置5min,低速瞬离5s以去除管盖内壁的液滴。
[0091]
(7)将上一步所得溶液吸取650μl到一个吸附柱中(吸附柱放入收集管中),8000rpm离心30sec,弃废液,将吸附柱放到一个新的2ml离心管中;
[0092]
(8)加入500ul缓冲液gd(使用前请先检查是否已加入无水乙醇),8000rpm离心30s,弃废液;
[0093]
(9)加入500ul缓冲液pw(使用前请先检查是否已加入无水乙醇),8000rpm离心30s,弃废液;
[0094]
(10)重复步骤(9)一次;
[0095]
(11)将吸附柱放入新2ml收集管中,12,000rpm(~13,400
×
g)离心2min,将吸附柱开盖置于事先准备好干净的1.5ml离心管中,室温干燥3min;
[0096]
(12)上述步骤(11)室温干燥目的是将缓冲液中乙醇去除干净,若乙醇有残留会影响后续的酶反应(酶切、酶连反应)实验;
[0097]
(13)向吸附膜中间位置悬空滴加50μl洗脱液,室温放置5min,12,000rpm(~13,400
×
g)离心2min,将溶液收集到离心管中;
[0098]
(14)使用dsdna hs assay kit for对提取的核酸dna(样品a)进行定量,定量结果如下所示:
[0099]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品a36.8501840
[0100]
制备例2
[0101]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0102]
(1)样品前处理i:取样品0.8ml,加入10μl溶菌酶(10mg/ml)和10μl溶壁酶(10000u/ml),混匀,37℃温育20min;
[0103]
(2)样品前处理ii:不进行物理破壁处理;
[0104]
(3)提取步骤(4)-(13)参见方案a;
[0105]
(4)使用dsdna hs assay kit for对提取的核酸dna(样品b)进行定量,定量结果如下所示:
[0106]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品b29.4501470
[0107]
从上述结果不难看出,不进行物理破壁处理,所提到的样品的浓度和总量均小于样品a。
[0108]
制备例3
[0109]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0110]
(1)样品前处理i:取样品0.8ml,加入10μl溶壁酶(10000u/ml),混匀,37℃温育
20min;
[0111]
(2)样品前处理ii:再加入200mg玻璃珠,使用均质仪高速震荡处理,其均质仪参数可设置为:60hz的速度振荡1min,间隔30s,共5个循环;
[0112]
(3)提取步骤(4)-(13)参见方案a;
[0113]
(4)使用dsdnahs assay kit for对提取的核酸dna(样品c)进行定量,定量结果如下所示:
[0114]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品c32.6501630
[0115]
从上述结果可以看出,仅采用溶壁酶进行处理,所提到的样品总量小于样品a。
[0116]
制备例4
[0117]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0118]
(1)样品前处理i:取样品0.8ml,加入10μl溶菌酶(10mg/ml),混匀,37℃温育20min;
[0119]
(2)样品前处理ii:再加入200mg玻璃珠,使用均质仪高速震荡处理,其均质仪参数可设置为:60hz的速度振荡1min,间隔30s,共5个循环);
[0120]
(3)提取步骤(4)-(13)参见方案a;
[0121]
(4)使用dsdna hs assay kit for对提取的核酸dna(样品d)进行定量,定量结果如下所示:
[0122]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品d30.8501540
[0123]
从上述结果可以看出,仅采用溶菌酶进行处理,所提到的样品总量小于样品a。
[0124]
制备例5
[0125]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0126]
(1)样品前处理i:不进行酶反应破壁处理;
[0127]
(2)样品前处理ii:取样品0.8ml,再加入200mg玻璃珠,使用均质仪高速震荡处理,其均质仪参数可设置为:60hz的速度振荡1min,间隔30s,共5个循环);
[0128]
(3)提取步骤(4)-(13)参见方案a;
[0129]
(4)使用dsdna hs assay kit for对提取的核酸dna(样品e)进行定量,定量结果如下:
[0130]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品e28501400
[0131]
从上述结果可以看出,仅进行高速震荡处理,所提到的样品总量以及样品浓度小于样品a。
[0132]
制备例6
[0133]
利用上述微生物细胞样品进行如下实验,具体操作步骤如下:
[0134]
(1)样品前处理i:不进行酶反应破壁处理;
[0135]
(2)样品前处理ii:不进行物理破壁处理;
[0136]
(3)提取步骤:取样品0.6ml,置新2ml离心管中;后续提取步骤(4)-(13)参见方案
a;
[0137]
(4)使用dsdna hs assay kit for对提取的核酸dna(样品f)进行定量,定量结果如下:
[0138]
样品编号浓度(ng/ul)体积(ul)总量(ng)样品f24.8501240
[0139]
从上述结果可以看出,既不进行酶解破壁处理,也不进行物理破壁处理,所提到的样品总量还有样品浓度均远小于样品a。
[0140]
将制备例1-6中,提取的dna用q-pcr方法检测酿酒酵母、新型隐球菌和金黄色葡萄球菌含量,并记录检测的ct值如下表2所示:
[0141]
表2不同样品的ct值结果
[0142][0143]
根据上表实验结果,增加使用溶菌酶、溶壁酶和均质仪物理破壁联合处理样本后,提取得到的dna样品,酿酒酵母、隐球菌、金黄色葡萄球菌检测的ct值比较小;所检测阳性样本的含量最高。因此,在一般以化学试剂裂解细胞的基础上,增加使用酶解方法与物理破壁方法结合对微生物细胞进行细胞壁破碎处理,能够高效的破碎真菌、结核类杆菌、革兰氏阳性菌的细胞壁,提高这类病原体的检测灵敏度;
[0144]
实施例2
[0145]
实施例2以真实呼吸道感染样品为实验样本,包含肺泡灌洗液、痰液等,样品信息如下表3所示:
[0146]
表3不同样品类型以及编号
[0147]
样品编号样品类型样品体积jx肺泡灌洗液5mlymm肺泡灌洗液5mlnlf肺泡灌洗液5mltxx肺泡灌洗液5mlxsh肺泡灌洗液5mlzhf痰液2mlwwk痰液2ml
xsk痰液2mlzl痰液2mlyzd痰液2ml
[0148]
然后按照下述方法对样品进行处理,并提取核酸:
[0149]
(1)样品前处理i:取不同感染样品0.8ml,加入10μl溶菌酶(10mg/ml)和10μl溶壁酶(10000u/ml),混匀,37℃温育20min;
[0150]
(2)样品前处理ii:再加入200mg玻璃珠,使用均质仪高速震荡处理,其均质仪参数可设置为:60hz的速度振荡1min,间隔30s,共5个循环);
[0151]
(3)8000rpm离心30s,转移上清600ul置新2ml离心管中;
[0152]
(4)向2ml离心管中加入2ul carrier rna(1ug/ul)储存液,60ul蛋白酶k和600ul缓冲液gb,震荡混合15s,低速瞬离5s以去除管盖内壁的液滴;
[0153]
(5)在65℃温育15min,并不时摇动样品。低速瞬离5s以去除管盖内壁的液滴;
[0154]
(6)待样品温度降至室温后加入600μl预冷的无水乙醇,并涡旋混匀15s,室温放置5min,低速瞬离5s以去除管盖内壁的液滴。
[0155]
(7)将上一步所得溶液吸取650μl到一个吸附柱中(吸附柱放入收集管中),8000rpm离心30sec,弃废液,将吸附柱放到一个新的2ml离心管中;
[0156]
(8)加入500ul缓冲液gd(使用前请先检查是否已加入无水乙醇),8000rpm离心30s,弃废液;
[0157]
(9)加入500ul缓冲液pw(使用前请先检查是否已加入无水乙醇),8000rpm离心30s,弃废液;
[0158]
(10)重复步骤(9)一次;
[0159]
(11)将吸附柱放入新2ml收集管中,12,000rpm(~13,400
×
g)离心2min,将吸附柱开盖置于事先准备好干净的1.5ml离心管中,室温干燥3min;
[0160]
(12)上述步骤(11)室温干燥目的是将缓冲液中乙醇去除干净,若乙醇有残留会影响后续的酶反应(酶切、酶连反应)实验;
[0161]
(13)向吸附膜中间位置悬空滴加60μl洗脱液,室温放置5min,12,000rpm(~13,400
×
g)离心2min,将溶液收集到离心管中;
[0162]
(14)使用dsdna hs assay kit for对提取的核酸dna进行定量,定量结果如下表4所示:
[0163]
表4定量结果
[0164]
样品编号dna浓度(ng/ul)体积(ul)总量(ng)jx4.7160282.6ymm7.1360427.8nlf26.6601596txx15.860948xsh8.4160504.6zhf3.4660207.6wwk1.8260109.2xsk10.760642
zl8.1960491.4yzd8.2260493.2
[0165]
然后参照如下方法构建高通量测序文库,包括:
[0166]
(1)利用片段化酶(dna smearase mix,厂家:翌圣生物科技(上海)股份有限公司)对提取得到的dna样品进行片段化处理,每个dna样品取用100ng。具体的,将片段化酶解冻后颠倒混匀,于灭菌200ul pcr管中配制如下反应:
[0167]
试剂体积dna smearase mix10uldnax ulddh2o50-x ul
[0168]
使用移液器轻轻吹打20次充分混匀,并短暂离心将反应液收集至管底。在pcr仪中运行反应程序,程序运行到4℃时,按暂停键,将含反应体系的pcr管放入pcr仪,然后继续运行程序,反应程序:105℃,热盖;4℃,1min;30℃,15min;72℃,20min;4℃,hold;
[0169]
(2)使用连接酶反应体系,将退火y型全长unique dual index(udi)的接头序列连接在步骤(1)中获得的dna片段两端,所述接头序列上含8nt唯一样本双标签(index);具体的,将连接缓冲液解冻后颠倒混匀在步骤(1)中获得的反应产物pcr管中配制如下反应:
[0170]
试剂体积第(1)步产物60uldna adapter no.x5ulligation enhancer30ulquick t4 dna ligase5ul
[0171]
no.x为index编号,每个样品对应一个的unique dual index编号,使用移液器轻轻吹打20次充分混匀,并短暂离心将反应液收集至管底。在pcr仪中运行反应程序,反应程序:20℃,15min;4℃,hold。
[0172]
(3)对步骤(2)中的产物通过羧基磁珠进行纯化,获得高通量测序文库。所述羧基磁珠为hieffdna selection beads。
[0173]
所述磁珠纯化步骤为:
[0174]
将磁珠提前30min拿出涡旋震荡混匀,平衡至室温,磁珠平衡至室温后,涡旋震荡混匀;在1.5ml离心管中,吸取60ul至100ul接头连接反应产物中,涡旋振荡或使用移液器轻轻吹打10次充分混匀;室温孵育5min,然后将1.5ml离心管短暂离心并置于磁力架中分离磁珠和液体,待溶液澄清后(放置约5min),小心移除上清;保持1.5ml离心管置于磁力架中,加入200ul新鲜配制的80%乙醇漂洗磁珠,将1.5ml离心管在磁力架上小心缓慢旋转180度,室温孵育30s,再将1.5ml离心管在磁力架上小心缓慢旋转180度,室温孵育30s,待磁珠完全移动到贴近磁条的管壁上,小心移除上清;重复漂洗步骤一次,总计漂洗两次;保持1.5ml离心管始终置于磁力架中,打开离心管管盖,空气干燥磁珠5-10min至无乙醇残留;将1.5ml离心管从磁力架中取出,加入31ul ddh2o洗脱,涡旋振荡混匀,于室温静置5min,将1.5ml离心管短暂离心并置于磁力架上静置,待溶液澄清后(放置约5min),小心吸取28ul上清至新的1.5ml离心管中,切勿触碰磁珠,即得纯化产物(测序文库)。
[0175]
使用dsdna hs assay kit for和agilent dna 1000 kit试剂对测序文库进行浓度和片段大小检测,并记录检测结果如下表5:
[0176]
表5测序文库检测结果
[0177]
样品编号文库浓度(ng/ul)片段大小(bp)总量(ng)jx1.3840038.64ymm1.7841049.84nlf1.9142253.48txx1.9841855.44xsh1.7248848.16zhf1.1646032.48wwk6.76412189.28xsk1.4242439.76zl5.5427154yzd3.87410108.36
[0178]
将定量后的测序文库混合成上机文库,对上机测序文库进行片段大小和浓度检测,稀释至0.5-3.5pm,在测序仪上进行高通量测序,具体步骤如下:
[0179]
将测序文库按照每个20m数据量进行混合成上机文库,上机测序文库稀释成1.2pm,使用nextseq 500/550高通量v2试剂盒(75次循环),按照其使用说明书,在illumina nextseq dx测序仪上进行高通量测序;
[0180]
对获得的测序数据进行分析,具体步骤如下:
[0181]
根据每个样本在文库构建过程中所添加的特异双index分子序列,将属于每个样本的测序reads数据进行拆分,拆分后的reads数据通过生物信息学分析软件,首先评价其数据质量,too_short_filter、dupliation、q30值等,将每个测序文库的reads序列信息与人源数据库比对,计算数据中的人源序列比例,然后将比对到的人源序列剔除,得到不含人源序列的reads;
[0182]
表6测定结果
[0183]
[0184]
将不含人源序列的reads序列与微生物数据库比对,并通过分析,获得样本含有的病原微生物物种信息,如下表7所示:
[0185]
表7
[0186]
[0187]
[0188][0189]
根据上表实验结果可知,采用本发明方法进行病原检测,其匹配的病原体reads数相比于现有技术,检测结果更为准确、可靠。
[0190]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
[0191]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0192]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1