一种成纤维细胞生长因子受体抑制剂的中间体及其制备方法和用途与流程
1.本发明属于药物合成领域,具体涉及一种成纤维细胞生长因子受体抑制剂的中间体及其制备方法和用途。
背景技术:
2.蛋白激酶为一类调节各种细胞功能的蛋白质(酶类)。这是通过在蛋白底物上特定氨基酸的磷酸化而使得底物蛋白构象改变来完成的。构象的变化调节底物活性或其与其他结合配偶体相互作用的能力。蛋白激酶的酶活性是指激酶往底物上添加磷酸根基团的速率。这可通过例如测定被转化为产物的底物的量与时间的函数来测定。在蛋白激酶的活性位出现底物的磷酸化。
3.酪氨酸激酶为在蛋白底物上催化三磷酸腺苷(atp)的末端磷酸转化为酪氨酸残基的蛋白激酶的亚组。这些激酶在致使细胞增殖、分化和迁移的生长因子信号传导的传播中具有重要作用。
4.成纤维细胞生长因子(fgf)被认为是许多生理过程(如发育和血管发生过程中形态发生)的重要介质。目前存在超过25种的已知fgf家族成员。成纤维细胞生长因子受体(fgfr)家族包括四个成员,其各由胞外配体结合区、单跨膜区和细胞内胞质蛋白酪氨酸激酶区组成。在fgf刺激下,fgfr发生二聚作用和转磷酸作用,这导致受体活化。受体的活化足以恢复和激活特定的下游信号配偶体,所述下游信号配偶体参与各种过程如细胞生长、细胞代谢和细胞存活的调节(综述于eswarakumar,v.p.等,cytokine&growth factor reviews 2005,16,第139-149页)。结果,fgf和fgfr有可能引起和/或促进肿瘤形成。
5.现在相当多的证据表明fgf信号传导与人类癌症直接有关。有报道在不同范围的肿瘤类型如膀胱、肾细胞和前列腺(及其他)肿瘤中各种fgf的表达增加。fgf还被描述为强有力的血管生成因子。还有报道fgfr在内皮细胞中表达。各种fgfr的激活突变与膀胱癌和多发性骨髓瘤(及其他)有关,同时有文献证明受体在前列腺和膀胱癌及其他上也有表达(综述于grose,r.等,cytokine&growth factorreviews 2005,16,第179-186页和kwabi-addo,b.等,endocrine-related cancer 2004,11,第709-724页)。由于这些原因,特别是由于靶向fgfr和/或fgf信号传导的治疗可直接影响肿瘤细胞和肿瘤血管发生,fgf信号传导系统为具有吸引力的治疗靶点。
6.2008年,阿斯利康(瑞典)有限公司(astrazeneca ab)在一篇专利申请wo2008075068a2中公开了靶向fgfr和/或fgf信号传导的化合物,其中最有代表性的化合物是实施例154化合物(azd4547),化学结构如下:
[0007][0008]
目前,azd4547的制备主要包括以下几种方法:
[0009]
(1)专利申请wo2008075068a1公开了一种制备方法,包括如下步骤:
[0010][0011]
在该制备方法中,以3-(3,5-二甲氧基苯基)丙酸乙酯为原料通过三步反应制备azd4547,其中第一步反应需要通过柱层析纯化,收率仅为42%;第二步反应需要回流反应24小时,水合肼在高温反应中容易发生爆炸,且水合肼属于高毒性、基因毒性试剂,直接高温反应对人和环境不友好;第三步反应也需柱层析纯化,制备azd4547的三步反应总收率仅为21.08%;因此,该制备方法多步反应需柱层析操作,安全性差,收率低,不适合工业化,无法解决药物可及性问题。
[0012]
(2)专利申请cn111072638a公开了另一种制备方法,包括如下步骤:
[0013][0014]
在该制备方法中,以3-(3,5-二甲氧基苯基)丙酸为起始原料,经五步反应制备得到azd4547,总收率为42.5%,在该制备方法中需要用到剧毒试剂氰乙酸乙酯和昂贵试剂钯碳、氯化亚锡、雷尼镍,也不适合工业化生产。
[0015]
(3)此外,专利申请wo2016137506a1公开了一种azd4547关键中间体3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺的制备方法,具体如下:
[0016][0017]
在该制备方法中,第一步反应采用乙醇回流反应,第二步反应溶剂用量大,且需在-78℃超低温反应,反应完成还需采用柱层析纯化,不适合工业化应用。
[0018]
综上所述,现有技术中公开的azd4547制备方法不适合工业化应用,无法解决药物可及性问题。因此,特别需要开发一种可工业化的制备方法,来满足azd4547临床研究以及药物上市的需求。
技术实现要素:
[0019]
本发明的目的在于提供一种成纤维细胞生长因子受体抑制剂的中间体及其制备方法和用途,以解决药物可及性问题,满足azd4547临床研究以及药物上市的需求。
[0020]
本发明第一方面提供一种式(a)化合物,即3-(3,5-二甲氧苯基)丙酸异丙酯:
[0021][0022]
本发明第二方面提供一种式(a)化合物的制备方法,包括如下步骤:
[0023][0024]
式(sm)化合物经酯化反应制备得到式(a)化合物。
[0025]
作为优选的方案,所述制备方法中,式(sm)化合物与异丙醇反应生成式(a)化合物,所述式(sm)化合物和异丙醇的质量体积比为1:(1~50)。
[0026]
作为进一步优选的方案,所述制备方法中,式(sm)化合物与异丙醇反应生成式(a)化合物,所述式(sm)化合物和异丙醇的质量体积比为1:(5~20)。
[0027]
作为更进一步优选的方案,所述制备方法中,式(sm)化合物与异丙醇反应生成式(a)化合物,所述式(sm)化合物和异丙醇的质量体积比为1:(5~10)。
[0028]
作为优选的方案,所述制备方法中,式(sm)化合物与异丙醇反应体系中加入
socl2,所述式(sm)化合物与socl2的投料摩尔比为1:(0.1~10)。
[0029]
作为进一步优选的方案,所述制备方法中,式(sm)化合物与异丙醇反应体系中加入socl2,所述式(sm)化合物与socl2的投料摩尔比为1:(0.5~2)。
[0030]
作为优选的方案,在所述制备方法中,反应在40℃~80℃进行。
[0031]
作为优选的方案,在所述制备方法中,反应在55℃~65℃进行。
[0032]
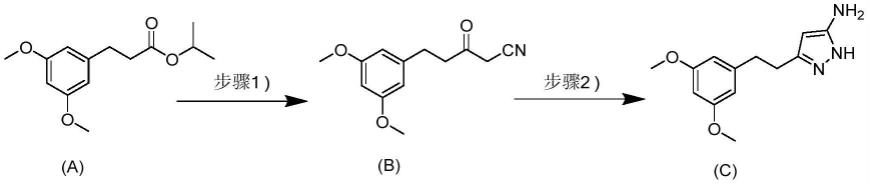
本发明第三方面提供一种式(a)化合物在制备式(c)化合物或其酸式盐中的用途,所述制备包括如下步骤:
[0033][0034]
1)式(a)化合物经反应制备得到式(b)化合物;
[0035]
2)式(b)化合物经反应制备得到式(c)化合物或其酸式盐。
[0036]
作为优选的方案,所述用途中,酸式盐为无机酸盐或者有机酸盐,所述无机酸盐选自盐酸盐、硫酸盐、氢溴酸盐、氢氟酸盐、氢碘酸盐或磷酸盐;所述有机酸盐选自醋酸盐、三氟乙酸盐、苯磺酸盐、对甲苯磺酸盐、4-氯苯磺酸盐、甲磺酸盐、乙磺酸盐、苯甲酸盐、柠檬酸盐、苹果酸盐、酒石酸盐、蚁酸盐、富马酸盐、半乳糖酸盐、丙二酸盐、乙醇酸盐、草酸盐、丙酸盐、4-乙酰氨基苯甲酸盐、4-氨基苯甲酸盐、水杨酸盐、4-氨基水杨酸盐、2,5-二羟基苯甲酸盐或1-羟基-2-萘甲酸盐。
[0037]
作为优选的方案,所述用途步骤1)中,式(a)化合物与乙腈反应生成式(b)化合物,所述式(a)化合物与乙腈的投料摩尔比为1:(1~50)。
[0038]
作为进一步优选的方案,所述用途步骤1)中,式(a)化合物与乙腈反应生成式(b)化合物,所述式(a)化合物与乙腈的投料摩尔比为1:(1~25)。
[0039]
作为更进一步优选的方案,所述用途步骤1)中,式(a)化合物与乙腈反应生成式(b)化合物,所述式(a)化合物与乙腈的投料摩尔比为1:(2~20)。
[0040]
作为优选的方案,所述用途步骤1)中,式(a)化合物与乙腈的反应体系中加入碱性试剂,所述碱性试剂选自二异丙基氨基锂、正丁基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或六甲基二硅基胺基锂中的一种或多种。
[0041]
作为进一步优选的方案,所述用途步骤1)中,所述式(a)化合物与碱性试剂的投料摩尔比为1:(1~20)。
[0042]
作为更进一步优选的方案,所述用途步骤1)中,所述式(a)化合物与碱性试剂的投料摩尔比为1:(2~8)。
[0043]
作为优选的方案,所述用途步骤1)中,式(a)化合物和乙腈的反应体系中加入的碱性试剂为二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-78℃~0℃。
[0044]
作为优选的方案,所述用途步骤1)中,式(a)化合物和乙腈的反应体系中加入的碱性试剂为二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-30℃~-10℃。
[0045]
作为优选的方案,所述用途步骤2)中,式(b)化合物与水合肼反应制备得到式(c)
化合物或其酸式盐,式(b)化合物和水合肼的投料摩尔比为1:(0.5~20)。
[0046]
作为进一步优选的方案,所述用途步骤2)中,式(b)化合物与水合肼反应制备得到式(c)化合物或其酸式盐,式(b)化合物和水合肼的投料摩尔比为1:(0.5~5)。
[0047]
作为优选的方案,所述用途步骤2)中,在酸性条件下反应,所述酸为无机酸或有机酸,所述无机酸选自盐酸、硫酸或磷酸;所述有机酸选自甲酸、乙酸、三氟乙酸、苯磺酸、对甲苯磺酸、甲磺酸、苯甲酸、富马酸、丙二酸、草酸或水杨酸。
[0048]
作为进一步优选的方案,所述用途步骤2)中,在乙酸条件下反应,所述式(b)化合物和乙酸的投料摩尔比为1:(0.5~20)。
[0049]
作为更进一步优选的方案,所述用途步骤2)中,在乙酸条件下反应,所述式(b)化合物和乙酸的投料摩尔比为1:(2~6)。
[0050]
作为进一步优选的方案,所述用途步骤2)中,先将水合肼和乙酸混合,再向上述混合液中加入式(b)化合物或其c
1-4
醇溶液或c
1-4
醇水溶液。
[0051]
作为进一步优选的方案,所述用途步骤2)中,反应在40℃~80℃下进行。
[0052]
作为进一步优选的方案,所述用途步骤2)中,反应在60℃~70℃下进行。
[0053]
本发明第四方面提供一种式(a)化合物在制备式(b)化合物中的用途,包括如下步骤:
[0054][0055]
式(a)化合物经反应制备得到式(b)化合物。
[0056]
作为优选的方案,所述用途中,式(a)化合物与乙腈反应生成式(b)化合物,所述式(a)化合物与乙腈的投料摩尔比为1:(2~20)。
[0057]
作为进一步优选的方案,所述用途中,式(a)化合物与乙腈的反应体系中加入碱性试剂,所述碱性试剂选自二异丙基氨基锂、正丁基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或六甲基二硅基胺基锂中的一种或多种。
[0058]
作为进一步优选的方案,所述用途中,所述式(a)化合物与碱性试剂的投料摩尔比为1:(2~8)。
[0059]
作为进一步优选的方案,所述用途中,式(a)化合物和乙腈的反应体系中加入的碱性试剂为二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-30℃~-10℃。
[0060]
本发明第五方面提供一种式(b’)化合物:
[0061][0062]
本发明第六方面提供一种式(a)化合物在制备azd4547或其酸式盐中的用途,包括通过前述制备方法由式(a)化合物制备得到式(c)化合物或其酸式盐,进一步还包括如下步骤:
[0063][0064]
式(c)化合物或其酸式盐与式(d)化合物反应得到azd4547或其酸式盐,
[0065]
其中r为c
1-8
烷基,优选为甲基、乙基或异丙基;所述酸式盐为无机酸盐或者有机酸盐;所述无机酸盐选自盐酸盐、硫酸盐、氢溴酸盐、氢氟酸盐、氢碘酸盐或磷酸盐;所述有机酸盐选自醋酸盐、三氟乙酸盐、苯磺酸盐、对甲苯磺酸盐、4-氯苯磺酸盐、甲磺酸盐、乙磺酸盐、苯甲酸盐、柠檬酸盐、苹果酸盐、酒石酸盐、蚁酸盐、富马酸盐、半乳糖酸盐、丙二酸盐、乙醇酸盐、草酸盐、丙酸盐、4-乙酰氨基苯甲酸盐、4-氨基苯甲酸盐、水杨酸盐、4-氨基水杨酸盐、2,5-二羟基苯甲酸盐或1-羟基-2-萘甲酸盐。
[0066]
作为优选的方案,所述用途中,式(c)化合物或其酸式盐与式(d)化合物在碱性试剂条件下缩合得到azd4547或其酸式盐,所述碱性试剂选自叔戊醇钾、叔丁醇钾、六甲基二硅基胺基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或二异丙基氨基锂中的一种或多种。
[0067]
本发明第七方面提供一种式(b)化合物的制备方法,包括如下步骤:
[0068][0069]
式(a)化合物在六甲基二硅基胺基钠、六甲基二硅基胺基锂、六甲基二硅基胺基钾或二异丙基氨基锂存在下与乙腈反应制备得到式(b)化合物。
[0070]
作为优选的方案,所述制备方法中,所述式(a)化合物与乙腈的投料摩尔比为1:(2~20)。
[0071]
作为进一步优选的方案,所述制备方法中,所述式(a)化合物与二异丙基氨基锂的投料摩尔比为1:(2~8)。
[0072]
作为进一步优选的方案,所述制备方法中,式(a)化合物和乙腈的反应体系中二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-30℃~-10℃。
[0073]
本发明第八方面提供一种式(c)化合物或其酸式盐的制备方法,包括如下步骤:
[0074][0075]
1)式(a)化合物在碱性条件下与乙腈反应制备得到式(b)化合物;
[0076]
2)式(b)化合物在酸性条件下与水合肼缩合反应生成式(c)化合物或其酸式盐;
[0077]
所述酸式盐为无机酸盐或者有机酸盐;所述无机酸盐选自盐酸盐、硫酸盐、氢溴酸盐、氢氟酸盐、氢碘酸盐或磷酸盐;所述有机酸盐选自醋酸盐、三氟乙酸盐、苯磺酸盐、对甲苯磺酸盐、4-氯苯磺酸盐、甲磺酸盐、乙磺酸盐、苯甲酸盐、柠檬酸盐、苹果酸盐、酒石酸盐、蚁酸盐、富马酸盐、半乳糖酸盐、丙二酸盐、乙醇酸盐、草酸盐、丙酸盐、4-乙酰氨基苯甲酸盐、4-氨基苯甲酸盐、水杨酸盐、4-氨基水杨酸盐、2,5-二羟基苯甲酸盐或1-羟基-2-萘甲酸盐。
[0078]
作为优选的方案,所述制备方法步骤1)中,式(a)化合物与乙腈的投料摩尔比为1:(2~20)。
[0079]
作为进一步优选的方案,所述制备方法步骤1)中,式(a)化合物与乙腈的反应体系中加入碱性试剂,所述碱性试剂选自二异丙基氨基锂、正丁基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或六甲基二硅基胺基锂中的一种或多种。
[0080]
作为进一步优选的方案,所述制备方法步骤1)中,所述式(a)化合物与碱性试剂的投料摩尔比为1:(2~8)。
[0081]
作为进一步优选的方案,所述制备方法步骤1)中,式(a)化合物和乙腈的反应体系中加入的碱性试剂为二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-30℃~-10℃。
[0082]
作为进一步优选的方案,所述制备方法步骤2)中,式(b)化合物和水合肼的投料摩尔比为1:(0.5~5)。
[0083]
作为进一步优选的方案,所述制备方法步骤2)在酸性条件下反应,所述酸为无机酸或有机酸,所述无机酸选自盐酸、硫酸或磷酸;所述有机酸选自甲酸、乙酸、三氟乙酸、苯磺酸、对甲苯磺酸、甲磺酸、苯甲酸、富马酸、丙二酸、草酸或水杨酸。
[0084]
作为进一步优选的方案,所述制备方法步骤2)中,在乙酸条件下反应,所述式(b)化合物和乙酸的投料摩尔比为1:(2~6)。
[0085]
作为进一步优选的方案,所述制备方法步骤2)中,先将水合肼和乙酸混合,再向上述混合液中加入式(b)化合物或其c
1-4
醇溶液或c
1-4
醇水溶液。
[0086]
作为进一步优选的方案,所述制备方法步骤2)中,反应在60℃~70℃下进行。
[0087]
本发明第九方面提供一种azd4547或其酸式盐的制备方法,包括如下步骤:
[0088][0089]
步骤1:式(sm)化合物在socl2存在下与异丙醇反应制备得到式(a)化合物;
[0090]
步骤2:式(a)化合物在碱性条件下与乙腈反应制备得到式(b)化合物;
[0091]
步骤3:式(b)化合物在酸性条件下与水合肼缩合反应生成式(c)化合物或其酸式盐;
[0092]
步骤4:式(c)化合物或其酸式盐与式(d)化合物反应得到azd4547或其酸式盐;
[0093]
其中r为c
1-8
烷基,优选为甲基、乙基或异丙基;所述步骤(3)或(4)中酸式盐为无机酸盐或者有机酸盐;所述无机酸盐选自盐酸盐、硫酸盐、氢溴酸盐、氢氟酸盐、氢碘酸盐或磷酸盐;所述有机酸盐选自醋酸盐、三氟乙酸盐、苯磺酸盐、对甲苯磺酸盐、4-氯苯磺酸盐、甲磺酸盐、乙磺酸盐、苯甲酸盐、柠檬酸盐、苹果酸盐、酒石酸盐、蚁酸盐、富马酸盐、半乳糖酸盐、丙二酸盐、乙醇酸盐、草酸盐、丙酸盐、4-乙酰氨基苯甲酸盐、4-氨基苯甲酸盐、水杨酸盐、4-氨基水杨酸盐、2,5-二羟基苯甲酸盐或1-羟基-2-萘甲酸盐。
[0094]
作为优选的方案,所述制备方法步骤1中,所述式(sm)化合物和异丙醇的质量体积比为1:(5~10)。
[0095]
作为优选的方案,所述制备方法步骤1中,式(sm)化合物与异丙醇反应体系中加入socl2,所述式(sm)化合物与socl2的投料摩尔比为1:(0.5~2)。
[0096]
作为进一步优选的方案,所述制备方法步骤1中,反应在55℃~65℃进行。
[0097]
作为优选的方案,所述制备方法步骤2中,式(a)化合物与乙腈的投料摩尔比为1:(2~20)。
[0098]
作为进一步优选的方案,所述制备方法步骤2中,式(a)化合物与乙腈的反应体系中加入碱性试剂,所述碱性试剂选自二异丙基氨基锂、正丁基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或六甲基二硅基胺基锂中的一种或多种。
[0099]
作为进一步优选的方案,所述制备方法步骤2中,所述式(a)化合物与碱性试剂的投料摩尔比为1:(2~8)。
[0100]
作为进一步优选的方案,所述制备方法步骤2中,式(a)化合物和乙腈的反应体系中加入的碱性试剂为二异丙基氨基锂,并且二异丙基氨基锂加入时控制反应体系温度为-30℃~-10℃。
[0101]
作为进一步优选的方案,所述制备方法步骤3中,式(b)化合物和水合肼的投料摩尔比为1:(0.5~5)。
[0102]
作为进一步优选的方案,所述制备方法步骤3在乙酸条件下反应,所述式(b)化合物和乙酸的投料摩尔比为1:(2~6)。
[0103]
作为进一步优选的方案,所述制备方法步骤3中先将水合肼和乙酸混合,再向上述混合液中加入式(b)化合物或其c
1-4
醇溶液或c
1-4
醇水溶液。
[0104]
作为进一步优选的方案,所述制备方法步骤3中先将水合肼和乙酸混合,再向上述混合液中加入式(b)化合物或其乙醇溶液或乙醇水溶液。
[0105]
作为进一步优选的方案,所述制备方法步骤3中,反应在60℃~70℃下进行。
[0106]
作为进一步优选的方案,所述制备方法步骤4中,式(c)化合物或其酸式盐与式(d)化合物在碱性试剂条件下缩合得到azd4547或其酸式盐,所述碱性试剂选自叔戊醇钾、叔丁醇钾、六甲基二硅基胺基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或二异丙基氨基锂中的一种或多种。
[0107]
本发明第十方面提供一种azd4547或其酸式盐的制备方法,包括如下步骤:
[0108][0109]
式(c)化合物或其酸式盐与式(d)化合物在叔戊醇钾、叔丁醇钾、六甲基二硅基胺基锂、六甲基二硅基胺基钠、六甲基二硅基胺基钾或二异丙基氨基锂存在下缩合得到azd4547或其酸式盐。
[0110]
本发明与现有技术相比具有如下优点:
[0111]
(1)本发明设计采用异丙酯结构,提供了一种新的式(a)化合物,即3-(3,5-二甲氧苯基)丙酸异丙酯,该酯化合物结构稳定,在工业化生产过程中可使得反应液均相澄清,可以有效避免采用甲酯式(a’)化合物或乙酯式(a”)化合物为反应原料时反应过程中出现粘胶团现象,导致反应过程中无法有效搅拌,使得所述制备方法无法实现工业化的问题。
[0112]
(2)本发明中实施例3或4中采用乙醇来淬灭反应,可以最大程度的避免反应液中的式(b)化合物产生自聚,影响后处理难度和产物纯度。
[0113]
(3)本发明实施例5步骤2采用在酸性条件下进行反应,尤其是乙酸,可以中和反应中的水合肼,使得反应过程更加安全,或者中和上步反应液中保留的碱性试剂。
[0114]
(4)本发明制备方法反应条件温和,避免超低温反应,且避免使用到柱层析纯化,溶剂用量少,收率高,可操作性强,可实现工业化应用。
附图说明
[0115]
图1是对比实验二中实验2反应中控hplc图谱。
[0116]
图2是实施例2有机相hplc谱图。
[0117]
图3是实施例2含杂质母液的hplc图谱,杂质化合物出峰时间为14.084min。
[0118]
图4是实施例2杂质的lcms图谱,杂质化合物的出峰时间为2.284min,对应于hplc图谱上14.084min的峰。
[0119]
图5是实施例2杂质的1hnmr图谱。
具体实施方式
[0120]
本技术的发明人经过广泛而深入地研究,设计提供了一种新的azd4547中间体,该azd4547中间体名称为3-(3,5-二甲氧苯基)丙酸异丙酯,并进一步公开了以该中间体为原料经两步反应制备得到azd4547关键中间体3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺,以及公开了上述两个中间体及其制备方法和在制备azd4547原料药(api)中的用途。本发明工艺成熟稳定、生产操作简单、收率高、成本低、安全环保,制备所得api可满足azd4547临床研究以及药物上市需求,解决了药物可及性问题。
[0121]
用在说明书和权利要求书中的下列术语具有下述含义。
[0122]
lda指二异丙基氨基锂,n-buli指正丁基锂,lihmds指六甲基二硅基胺基锂,nahmds指六甲基二硅基胺基钠,lioh指氢氧化锂,acoh指醋酸或乙酸,hcl指盐酸,thf指四氢呋喃,ko-tam指叔戊醇钾,tfa指三氟乙酸。eq为反应当量。
[0123]
下面结合实施例对本发明做进一步详细、完整地说明,但决非限制本发明,本发明也并非仅局限于实施例的内容。
[0124]
本发明的化合物结构是通过核磁共振(nmr)来确定的。nmr化学位移(δ)以百万分之一(ppm)的单位给出。
[0125]
nmr的测定是用bruker avance-400/500核磁仪,测定溶剂为氘代二甲基亚砜(dmso-d6),氘代甲醇(cd3od)和氘代氯仿(cdcl3),内标为四甲基硅烷(tms)。
[0126]
液质联用色谱lc-ms的测定用agilent technologies inifinitylab lc/msd质谱仪。
[0127]
hplc测定采用设备:agilent technologies 1260infinityⅱ,色谱柱:poroshell 120ec-c18 4μm 4.6
×
150mm,流动相:a相:水+0.05%三氟乙酸,b相:乙腈+0.05%三氟乙腈,流速:1.0ml/min。
[0128]
本发明实施例中的起始原料是已知的并且可以在市场上买到,或者可以采用或按照本领域已知的方法来合成。例如本领域普通技术人员可根据本领域公知的方法通过游离态物质与相应的酸反应制备获得本发明酸式盐。
[0129]
在无特殊说明的情况下,本发明的所有反应均在干燥氮气或氩气氛下进行,溶剂为干燥溶剂,反应温度单位为摄氏度(℃)。
[0130]
实施例1
[0131]
向1l的三口反应瓶中加入异丙醇(300ml),3-(3,5-二甲氧苯基)丙酸(60.0g,0.285mol),升温至40
±
5℃,搅拌5~10分钟使溶清。40
±
5℃下滴加socl2(37.3g,0.314mol),滴加时间≥0.5小时(滴加过程放热明显),滴毕,温度升高到60
±
5℃,搅拌反应
1小时,hplc检测原料反应完全。将反应液降至35
±
5℃,控温50℃以下减压浓缩至无明显馏分,加入甲基叔丁基醚(300ml)溶解,冰浴下加入5%碳酸钾水溶液调节反应液ph值到8~9,控温25
±
5℃搅拌0.5小时,静置分层,分出有机相,用饱和食盐水洗涤,控温45℃减压浓缩至干得到浅黄色油状物3-(3,5-二甲氧苯基)丙酸异丙酯72.1g,纯度:94%,收率:94.3%。
[0132]1hnmr(dmso-d6,400mhz)δ6.384-6.378(d,2h),6.318-6.306(t,1h),4.925-4.831(m,1h),3.706(s,6h),2.787-2.749(t,2h),2.571-2.533(t,2h),1.164-1.148(d,6h)。
[0133]
实施例2
[0134]
氮气保护下向500ml的三口反应瓶中加入3-(3,5-二甲氧苯基)丙酸异丙酯(20.0g,0.079mol),无水乙腈(80ml),无水四氢呋喃(100ml),将反应液降温至内温约-20℃,缓慢滴加二异丙基氨基锂(83ml,0.166mol,2m的thf溶液),约25分钟滴加完毕,继续搅拌5-10分钟,hplc检测原料反应完全,加入醋酸溶液(15ml)淬灭反应,减压浓缩,加入水(100ml),用25%碳酸钠水溶液调节ph至中性,加入乙酸乙酯(200ml)萃取(hplc图见图2),有机层减压浓缩至无馏分,加入乙醇(200ml)搅拌打浆,过滤,滤饼在45℃下真空干得到5-(3,5-二甲氧苯基)-3-氧代戊腈14.8g,纯度为98%,收率76%。
[0135]1hnmr(dmso-d6,400mhz)δ6.370-6.364(s,2h),6.320-6.309(s,1h),4.038(s,2h),3.709(s,6h),2.851-2.815(t,2h),2.739-2.702(t,2h)。
[0136]
经初步分离,含杂质母液hplc、杂质lcms和1hnmr图谱见图3-5。经解析,主要杂质由5-(3,5-二甲氧苯基)-3-氧代戊腈自聚产生,杂质化合物[式(b’)化合物]结构如下:
[0137][0138]
实施例3
[0139]
氮气保护下向500ml的三口反应瓶中加入3-(3,5-二甲氧苯基)丙酸异丙酯(11.29g,0.045mol),无水乙腈(40ml)和无水四氢呋喃(50ml),将反应液降温至-20℃,缓慢滴加二异丙基氨基锂四氢呋喃溶液(47ml,0.094mol),约25分钟滴加完毕,继续搅拌反应5-10分钟,hplc检测原料反应完全,加入无水乙醇(20ml)淬灭反应,再加入2-甲基四氢呋喃(50g)萃取,水层用盐酸调节ph至中性,过滤,滤饼在45℃下真空干燥得5-(3,5-二甲氧苯基)-3-氧代戊腈9.28g,纯度:99.5%,收率:88.0%。
[0140]
实施例4
[0141]
氮气保护下向3l的三口反应瓶中加入含3-(3,5-二甲氧苯基)丙酸异丙酯(120.0g,0.4756mol)的四氢呋喃溶液、无水乙腈(380g,9.25mol)、无水四氢呋喃(270g),搅拌,降至内温约-20℃并在此温度下,缓慢滴加二异丙基氨基锂(500ml,1mol)的四氢呋喃溶液,滴加完毕后约-20℃搅拌1-2小时,hplc检测原料转化完全,向反应中加入乙醇(474g)淬
灭反应,减压浓缩,加入纯化水,控制内温0-15℃,缓慢加入hcl,调节ph≈7.0,并析出大量固体,搅拌30min过滤,依次用纯化水、乙醇漂洗,45℃真空干燥得5-(3,5-二甲氧苯基)-3-氧代戊腈98.7g,纯度:99.6%,收率:89.0%。
[0142]
另外,发明人考察了该步反应中反应原料、无水乙腈、碱性试剂、反应温度对该反应的影响,hplc检测中控纯度和反应现象分别如下:
[0143]
[0144][0145]
由以上试验考察因素及试验现象可知,不同反应原料酯基基团种类、乙腈用量和碱性试剂对反应的影响:
[0146]
(1)反应原料酯基基团种类对反应的影响
[0147]
当反应原料选用具有甲酯基团的式(a’)化合物时,反应中均会形成粘胶团,影响搅拌,反应中控纯度不高。具体体现为:反应初始,反应液中出现粘胶团,影响搅拌,随着反应的进行,反应液逐渐胶粘化,甚至出现粘壁现象,无法搅拌。
[0148]
当反应原料选用具有乙酯基团的式(a”)化合物,反应中控纯度提高至87%,但反应中仍会形成粘胶团,影响搅拌,具体情况与反应原料选用具有甲酯基团的式(a’)化合物类似。
[0149]
在工艺放大过程中出现粘胶团容易导致反应不完全,甚至会出现缠绕搅拌桨,烧坏电机等危险情况,因此,上述两种制备工艺不适宜工业化放大生产。
[0150]
将反应原料的酯基结构改为异丙酯时,反应液均相澄清,无粘胶团现象,反应中控纯度提升至97%以上,适合工业化放大生产。发明人分析上述试验现象可能是由于异丙酯结构的稳定性较高,减少了副反应的形成。
[0151]
(2)乙腈用量对反应的影响
[0152]
本发明实验中的实验6和实验3,乙腈与反应原料的投料摩尔比从10eq上升至20eq时,反应中控纯度从90.4%上升至97.2%。
[0153]
对比实验一中实验2和实验3,乙腈与反应原料的投料摩尔比从1.2eq上升至25eq时,反应中控纯度从60.8%上升至92.8%。
[0154]
(3)碱性试剂的选用及用量对反应的影响
[0155]
从上述考察实验结果可以看出nahmds、lda和n-buli时反应中控纯度均较高。
[0156]
碱性试剂与反应原料的最佳投料摩尔比为2.1eq,低于2eq可能导致反应不完全。
[0157]
实施例5
[0158]
步骤1:5-(3,5-二甲氧苯基)-3-氧代戊腈(式(b)化合物)的合成
[0159]
氮气保护下向500ml的三口反应瓶中加入3-(3,5-二甲氧苯基)丙酸异丙酯
(20.0g,0.079mol),无水乙腈(80ml),无水四氢呋喃(100ml),将反应液降温至-20℃,缓慢滴加二异丙基氨基锂(83ml,0.166mol,2m的thf溶液),约25min加毕,搅拌反应5-10分钟,hplc检测原料反应完全,加入无水乙醇(40ml),减压浓缩至粘稠状,加入无水乙醇(60ml)制备成乙醇溶液,直接投入下一步反应。
[0160]
步骤2:3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺(式(c)化合物)的制备
[0161]
向500ml的三口反应瓶中加入乙酸(26.0g,0.436mol),乙醇(100ml),80%水合肼(15.0g,0.238mol),加热至内温约68℃,将步骤1得到的产物乙醇溶液(18.5g,0.079mol)在此温度下缓慢加入乙酸和水合肼混合溶液中,约40min加毕,加完在内温约68℃搅拌反应1小时,hplc检测5-(3,5-二甲氧苯基)-3-氧代戊腈转化完全;反应液减压浓缩,加入水(100ml),乙酸乙酯(200ml),加入约25%na2co3(40ml)调水层ph=7~8;分去水层,饱和食盐水(20ml)洗涤分层,减压浓缩有机层至无馏分,加入醋酸异丙酯(100ml)减压带至粘稠状,加入醋酸异丙酯(120ml)加热溶清,降温结晶,约10℃过滤,50℃真空干燥得3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺16.3g,纯度:99.6%,两步总收率:83%。
[0162]1hnmr(dmso-d6,400mhz)δ6.370-6.364(s,2h),6.320-6.309(s,1h),4.038(s,2h),3.709(s,6h),2.851-2.815(t,2h),2.739-2.702(t,2h)。
[0163]
另外,发明人考察了该步反应中乙酸用量对该反应的影响,hplc检测中控纯度如下:
[0164][0165]
另外,发明人还考察了将步骤1产物纯化后得到的固体式(b)化合物在乙酸存在下与水合肼反应,式(b)化合物也能转化完全,获得高纯式(c)化合物。
[0166]
实施例6
[0167]
向反应釜中加入3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺(100.0g,0.4044mol)、乙基4-((3r,5s)-3,5-二甲基哌嗪-1-基)苯酸酯(132.5g,0.5050mol)、2-甲基四氢呋喃(1300ml),加热至50~55℃搅拌1小时,经硅藻土过滤,滤液加入干净的反应釜中,加热常压蒸馏带水,将反应内温控制在78℃-88℃,缓慢滴加25%ko-tam甲苯溶液(490.0g),滴加时间约2小时,滴加完毕后调节反应内温在83-88℃搅拌3-6小时,取样检测原料反应完毕,将反应体系降温至30-60℃,缓慢加入水(8ml)淬灭反应,并在30-60℃搅拌0.5小时,再降温至约25℃,加入水(400ml),搅拌静置分层,分出有机相,加入水(200ml),加热至约50℃搅拌0.5小时,分去水层,如此重复2-3次,直至水层ph=7.0-9.5;有机层减压浓缩掉部分溶剂,残余物加热至80-90℃并搅拌1小时,缓慢降温至20-30℃,继续搅拌2-5小时,过滤,乙酸乙酯漂洗两次,45℃真空干燥得白色无定形固体状产物(azd4547)155.6g,纯度:98.5%,收
率:83%。
[0168]1hnmr(dmso-d6,400mhz)δ12.067(s,1h),10.275(s,1h),7.888-7.867(d,2h),6.943-6.922(d,2h),6.437-6.409(m,3h),6.317(s,1h),3.712-3.692(m,8h),2.861-2.803(m,6h),2.230-2.174(m,3h),1.036-1.020(d,6h)。
[0169]
实施例7
[0170]
向反应瓶中加入3-(3,5-二甲氧基苯乙基)-1h-吡唑-5-胺(10.0g,0.040mol)、乙基4-((3r,5s)-3,5-二甲基哌嗪-1-基)苯酸酯(12.1g,0.047mol),无水四氢呋喃(170ml),加热常压蒸馏至剩余约100ml,降温至-30℃~-20℃,缓慢滴加nahmds(0.125mol,63ml,2m的thf溶液),控制反应体系温度约-25℃搅拌20分钟,hplc检测原料基本反应完全,控温下缓慢加入水(30ml)淬灭,再加入冰醋酸(约10ml)中和,升温至约0℃搅拌,加入20%na2co3(10ml),减压浓缩至无馏份,在残余物中加入乙酸乙酯(120ml),加热至约45℃搅拌分层,分出有机相,加入饱和食盐水(30ml)洗涤一次,减压浓缩有机层至剩余约(30ml),再加入乙酸乙酯(30ml),再减压浓缩,如此重复两次,析出大量固体,补加乙酸乙酯至物料体积约50ml,控温0-10℃下搅拌1小时,过滤,45℃真空干燥得白色无定形固体状产物(azd4547)16.87g,纯度99.8%,收率:91%。
[0171]
在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述描述内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本技术所附权利要求书所限定的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1