一种用于多基因转化的慢病毒载体表达系统的制作方法
1.本发明属于生物医学领域,具体涉及一种利用分裂单一筛选标记实现对多基因转化的慢病毒载体表达系统,包括慢病毒载体表达系统、其构建方法以及在多基因转化中的应用。
背景技术:
2.慢病毒载体(lentiviral vector)是在hiv
‑
1病毒基础上改造而成的病毒载体系统,它能高效的将目的基因导入动物和人的原代细胞或细胞系。慢病毒载体介导的基因表达持续且稳定,原因是插入基因整合到宿主细胞基因组中,并随细胞基因组的分裂而分裂。另外,慢病毒有广泛的宿主,可感染分裂和不分裂的细胞,几乎可以感染所有类型的细胞,特别适合质粒载体转染效率低的细胞。
3.目前常用的慢病毒表达载体多是采用双启动子的表达元件串联的,同时具有抗生素抗性基因和荧光蛋白双重筛选基因,用于选择目的基因型的工程细胞,但是选择有限。只有几种有限数量的具有良好特征的抗生素耐药基因可用于真核细胞,同样也只有几种限数量的荧光蛋白的光谱可通过常用设备明确区分。如果研究人员要将多个基因转入一个细胞,他们经常会遇到没有足够的可选择标记的问题。另一方面,同时使用多种抗生素进行选择,对细胞往往是严酷的。
4.2006年,山中伸弥的团队发明了一种由oct4,sox2,klf4和c
‑
myc四种转录因子构成的“鸡尾酒”法,能够成功将终端分化的皮肤成纤维细胞重编程成为具有分化多能性的干细胞。这一突破性的发明突破了在医学上使用人胚胎干细胞的伦理限制,大大拓展了干细胞技术在临床医学上的应用潜力。但是这四个基因同时在一个慢病毒载体表达时,慢病毒的包装效率与这四个基因的表达都比较低,如何将这些具有广泛应用前景的目的基因进行有效包装,提高基因的表达率是我们亟待解决的问题。
技术实现要素:
5.为解决上述问题,让多个基因在一种筛选条件下被选择,本发明提供一种慢病毒载体表达系统、其构建方法与应用,具体的:本发明第一方面,提供一种慢病毒载体表达系统,所述慢病毒载体表达系统包括两个载体,其中第一个慢病毒载体包括n
‑
screening marker
‑
ts1片段,所述n
‑
screening marker
‑
ts1片段包括编码筛选标记蛋白的n端氨基酸的标记基因5’端核苷酸序列(n
‑
screening marker)和剪切接头1(ts1),第二个慢病毒载体包括ts2
‑
c
‑
screening marker片段,所述ts2
‑
c
‑
screening marker片段包括剪切接头2(ts2)和编码筛选标记蛋白的c端氨基酸的标记基因3’端核苷酸序列(c
‑
screening marker),所述两个慢病毒载体中的筛选标记蛋白相同,所述筛选标记蛋白的n端氨基酸和c端氨基酸连接后具有筛选标记功能。
6.优选的,所述标记基因包括选择基因和报告基因,优选的,所述选择基因包括抗生素抗性基因,更优选的,所述抗生素抗性基因包括但不仅限于新霉素磷酸转移酶基因
(npt),卡那霉素抗性基因(nptii),嘌呤霉素抗性基因(puro),四环素抗性基因,青霉素抗性基因,链霉素抗性基因等等;所述报告基因包括氯霉素乙酰转移酶 (cat),β
‑
半乳糖苷酶基因(lacz),二氢叶酸还原酶基因,人生长激素(hgh),分泌型碱性磷酸酶(seap),荧光素酶(luciferase),荧光蛋白等。
7.本发明的一个具体实施方式中,上述慢病毒载体系统中,基于病毒rna的剪接顺式元件,将筛选标记分成n端和c端两部分,只有当两个慢病毒载体导入同一个细胞,并且转录出相应的筛选标记的n端和c端rna时,在剪接顺式元件ts1,ts2作用下,会形成一个完整的筛选标记rna,进一步表达筛选标记蛋白。
8.在一个具体的实施方案中,本发明提供一种慢病毒表达系统,所述包括慢病毒表达系统两个慢病毒载体,其中第一个慢病毒载体包括plent
‑
ef1a
‑
mcs
‑
cmv
‑
n
‑
screening marker
‑
ts1的核苷酸序列,优选的,所述第一个慢病毒载体的核苷酸序列如seq id no.1所示。
9.第二个慢病毒表达载体包括plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑
screening marker的核苷酸序列,优选的,所述第二个慢病毒表达载体的核苷酸序列如seq id no.2所示。
10.进一步优选的,其中,kozak序列的核苷酸序列信息为gccacc;所述筛选标记基因为嘌呤霉素抗性基因(puro),编码n
‑ꢀ
puro 的dna序列如seq id no.3所示;tans splicing 1(ts1)序列如seq id no.4所示;tans splicing 2(ts2)序列如seq id no.5所示;编码c
‑ꢀ
puro的dna序列如seq id no.6所示。优选的,所述第一个慢病毒载体和第二个慢病毒载体还包括多个目的基因。更优选的,所述多个包括至少两个,例如2个,3个,4个,5个,6个或更多。
11.更优选的,所述目的基因包括oct4,sox2,klf4,c
‑
myc,nanog和lin28。
12.优选的,所述目的基因分别插入到第一个慢病毒载体和第二个慢病毒载体中。
13.更优选的,所述第一个慢病毒载体plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
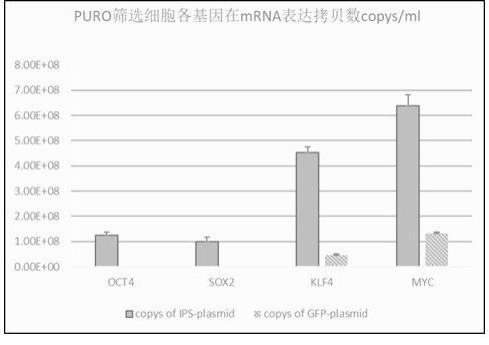
n
‑
puro
‑
ts1,所述第二个慢病毒载体包括plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro。
14.进一步优选的,oct4
‑
p2a
‑
sox2核苷酸序列如seq id:no.7所示,klf4
‑
p2a
‑
myc核苷酸序列如seq id no.8所示。
15.本发明第二方面,提供一种上述慢病毒载体表达系统的构建方法,其中所述构建方法包括:1)通过pcr技术获得n
‑
screening marker
‑
ts1片段和ts2
‑
c
‑
screening marker片段;2)将步骤1)获得的n
‑
screening marker
‑
ts1片段和ts2
‑
c
‑
screening marker片段分别插入第一慢病毒载体和第二慢病毒载体中获得包含标记基因和剪切接头的载体1和载体2。
16.在一个具体实施方式中,所述载体1为过表达载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
n
‑
screening marker
‑
ts1,所述载体2为过表达载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑
screening marker,所述构建方法的步骤1)和2)包括:s1:通过pcr技术获得n
‑
screening marker
‑
ts1片段,将其克隆进plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro载体中的puro处,获得载体1。
17.s2:通过pcr技术获得ts2
‑
c
‑
screening marker片段,将其克隆进plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro载体中的puro处,获得载体2。
18.进一步,其具体操作为:(1)设计引物(2)pcr通过pcr技术获得n
‑
puro
‑
ts1片段和ts2
‑
c
‑
puro片段,pcr反应结束后,取产物用电泳分离检测,若目的条带大小正确,利用并用pcr纯化回收试剂盒回收目的片段。
19.(3)酶切:(a)将pcr得到的n
‑
puro
‑
ts1,ts2
‑
c
‑
puro进行酶切;(b)加样混匀后置于37℃酶切2h,反应结束后用1.5%琼脂糖凝胶电泳检测酶切目的条带大小,并用胶回收试剂盒回收目的片段;(c)将载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro进行酶切,酶切体系如下表,然后胶回收载体;(d)连接:将回收的目的基因片段与经过同样双酶切的载体连接。
20.混匀后微离心,22℃连接1h。
21.(4)转化:连接产物转化大肠杆菌dh5α感受态细胞,涂布于相应抗性的lb平板上进行筛选;(a)从
‑
80℃取出提前制备好的dh5a感受态置于冰浴中。
22.(b)待dh5a感受态细胞融化后,取5μl连接产物于20μl dh5a感受态细胞中,充分混匀,冰浴中静置15分钟。
23.(c)将离心管放入42℃水浴锅中40秒(期间不要摇动离心管),然后快速移至冰浴中,静置2分钟。
24.(d)向离心管中加入200μl的无菌的lb培养基(不加抗生素),混匀后置于摇床中37℃,220rpm,振摇1小时。目的是使质粒上相关的抗性标记基因表达,使菌体复苏。
25.(e)涂布到相应抗性的固体培养基平皿中;(f)37℃培养箱中培养过夜。
26.(5)酶切验证;(6)测序:挑取单菌落培养后提取质粒用ecori/mssi双酶切进行酶切鉴定阳性克隆,并送测序验证。
27.优选的,所述构建方法还包括:3)将目的基因分别插入步骤2)获得的载体1和载体2中,获得载体3和载体4。
28.更优选的,所述目的基因为oct4,sox2,klf4和myc。
29.进一步优选的,所述载体3和载体4分别为plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1载体(p3)和plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro载体(p4)。
30.其中oct4
‑
p2a
‑
sox2核苷酸序列如seq id:no.7所示,klf4
‑
p2a
‑
myc核苷酸序列如seq id no.8所示。
31.本发明第三方面,提供一种上述任一的慢病毒载体表达载体在多个目的基因转化中的应用。
32.优选的,所述多个目的基因包括至少两个目的基因,例如2个,3个,4个,5个,6个或更多目的基因。
33.优选的,所述目的基因包括oct4,sox2,klf4,c
‑
myc,nanog和/或lin28两个或两个以上任意基因的组合。
34.本发明的慢病毒载体表达系统、其构建方法和用途的优点在于:1.本发明的慢病毒载体表达系统用单一的筛选标记可以实现多个目的基因的表达,例如oct4,sox2,klf4和myc基因的表达,将其转染细胞后,仅用puro抗生素抗性基因进行筛选,得到与gfp
‑
puro载体一致的存活率。
35.2.利用本发明的慢病毒载体表达系统可以提高目的蛋白的表达率,例如,两个cmv启动子分别启动oct4
‑
p2a
‑
sox2和klf4
‑
p2a
‑
myc相对于cmv启动子启动oct4
‑
p2a
‑
sox2
‑
t2a
‑
klf4
‑
p2a
‑
myc,各基因的表达量都显著提高。毕竟启动子的启动相率有限,离启动子越远的基因,表达量越低。
36.3.利用本发明的慢病毒载体表达系统可以在多个细胞中稳定表达目的蛋白,例如hek293,293a细胞等。
37.4.单一筛选标记可以避免多个筛选标记对细胞的损伤以及环境的污染,例如通常需要puro和blasticidin两个筛选标记对同一细胞进行两次筛选,利用本发明的慢病毒载体表达系统只需要利用一种筛选标记进行一次筛选。
附图说明
38.图1:为plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro
‑
ts1(p1)电泳检测结果,其中m为marker示意图,p1为实际marker条带,c为检测载体电泳条带。
39.图2:为plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑
puro(p2)电泳检测结果,其中m为marker示意图,p2为实际marker条带,c为检测载体电泳条带。
40.图3:为plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1(p3)电泳检测结果,其中m为marker示意图,p3为实际marker条带,c为检测载体电泳条带。
41.图4:为plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro(p4) 电泳检测结果,其中m为marker示意图,p4为实际marker条带,c为检测载体电泳条带。
42.图5:为48小时后转染结果,图5a为h2o+pei白光结果,图5b为h2o+pei 绿光结果;图5c为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pei白光结果,图5d为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pei绿光结果,图5e为gfp
‑
puro+pei白光结果,图5f为gfp
‑
puro+pei绿光结果。
43.图6:为puro筛选72小时后细胞生长状态。其中,图6a为h2o+pei的白光结果,图6b为h2o+pei的绿光结果,图6c为ips
‑
n
‑
puro +ips
‑
c
‑
puro+pe1的白光结果,图6d为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pe1的绿光结果,图6e为gfp
‑
puro+pei 白光结果,图6f为gfp
‑
puro+pei 绿光结果图7:为目的蛋白mrna在hek293细胞中表达拷贝数。
44.图8:为ips
‑
n
‑
puro
‑
moi 6+ips
‑
c
‑
puro
‑
moi 6组293a细胞的感染结果,图a和b分别为加压前和加压后的结果。
45.图9:为ips
‑
n
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果
图10:为ips
‑
c
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果图11:为lenti
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果图12:为空白对照组293a细胞的转染结果,图a和b分别为加压前和加压后的结果。
46.图13:为目的蛋白mrna在293a细胞中的表达拷贝数。
具体实施方式
47.下面结合具体实施例对本发明进行详细描述。但是,本发明可以以许多不同的形式来实现,并不限于本文所描述的实施例。相反地,提供这些实施例的目的是使对本发明的公开内容的理解更加透彻全面。
48.除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
49.在下述每一实施例中,主要的设备和材料是从以下所指出的几家公司获得:1. 实验材料表1:实验材料
2. 实验设备:表2:主要设备
3. 统计分析实验数据用均值
±
标准差(mean
±
sd)表示。两组数据用非配对t检验(non
‑
paired t test),以p<0.05为有显著性统计学意义。
50.实施例1:构建第一和第二慢病毒载体一、载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
n
‑
puro
‑
ts1和plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑ꢀ
puro的构建1. 设计引物。具体引物见表3:表3:pcr引物2. pcr,通过pcr技术获得n
‑
puro
‑
ts1片段和ts2
‑
c
‑
puro片段。
51.表4:pcr体系:(单位:μl)
反应程序:表5:pcr程序pcr反应结束后,取2
µ
lpcr产物用1.5%琼脂糖凝胶电泳分离检测,若目的条带大小正确,利用并用pcr纯化回收试剂盒回收目的片段。
52.3. 酶切(1)将pcr得到的n
‑
puro
‑
ts1,ts2
‑
c
‑
puro进行酶切,筛选基因酶切体系如表6:表6:筛选基因酶切体系(2)加样混匀后置于37℃酶切2h,反应结束后用1.5%琼脂糖凝胶电泳检测酶切目的条带大小,并用胶回收试剂盒回收目的片段。
53.(3)将载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro进行酶切,酶切体系如表7,然后胶回收载体。
54.表7:载体酶切体系
4. 连接将回收的筛选基因片段与经过同样双酶切的载体连接,连接体系如表8:表8:连接体系混匀后微离心,22℃连接1h。
55.5. 转化连接产物转化大肠杆菌dh5α感受态细胞,涂布于相应抗性的lb平板上进行筛选;(1)从
‑
80℃取出提前制备好的dh5a感受态置于冰浴中;(2)待dh5a感受态细胞融化后,取5μl连接产物于20μl dh5a感受态细胞中,充分混匀,冰浴中静置15分钟;(3)将离心管放入42℃水浴锅中40秒(期间不要摇动离心管),然后快速移至冰浴中,静置2分钟;(4)向离心管中加入200μl的无菌的lb培养基(不加抗生素),混匀后置于摇床中37℃,220rpm,振摇1小时;目的是使质粒上相关的抗性标记基因表达,使菌体复苏;(5)涂布到相应抗性的固体培养基平皿中;(6)37℃培养箱中培养过夜。
56.6. 酶切验证:反应体系如表9,用1.5%琼脂糖凝胶电泳检测酶切目的条带大小。
57.表9:酶切验证反应体系结果如图1和2所示:其中图1为plent
‑
ef1a
‑
mcs
‑
cmv
‑
puro
‑
ts1(p1)电泳检测结
果,得到520bp片段,表明获得具有筛选基因puro 5’端的目的片段,图2为plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑
puro(p2)电泳检测结果,得到320bp片段,表明获得具有筛选基因puro 3’端的目的片段。
58.7. 测序挑取单菌落培养后提取质粒用ecori/mssi双酶切进行酶切鉴定阳性克隆,并送测序验证。
59.二、构建plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1载体和plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro载体:1. 设计引物,见表10:表:10:pcr引物2. pcr表11:pcr体系:(单位:μl)反应程序:表12:pcr反应程序
pcr反应结束后,取2
µ
l pcr产物用1.5%琼脂糖凝胶电泳分离检测,若目的条带大小正确,利用并用pcr纯化回收试剂盒回收目的片段。
60.3. 酶切(1)将pcr得到的oct4
‑
p2a
‑
sox2与klf4
‑
p2a
‑
myc进行酶切,目的基因酶切体系如表13所示:表13:目的基因酶切体系(2)加样混匀后置于37℃酶切2h,反应结束后用1.5%琼脂糖凝胶电泳检测酶切目的条带大小,并用胶回收试剂盒回收目的片段。
61.(3)将载体plent
‑
ef1a
‑
mcs
‑
cmv
‑
n
‑
puro
‑
ts1和plent
‑
ef1a
‑
mcs
‑
cmv
‑
ts2
‑
c
‑
puro进行酶切,酶切体系如表14,然后胶回收载体。
62.表14:载体酶切体系
4. 连接将回收的目的基因片段与经过同样双酶切的载体连接,连接体系如表15所示:表15:连接体系混匀后微离心,22℃连接1h。
63.5. 转化连接产物转化大肠杆菌dh5α感受态细胞,涂布于相应抗性的lb平板上进行筛选;(1)从
‑
80℃取出提前制备好的dh5a感受态置于冰浴中;(2)待dh5a感受态细胞融化后,取5μl连接产物于20μl dh5a感受态细胞中,充分混匀,冰浴中静置15分钟;(3)将离心管放入42℃水浴锅中40秒(期间不要摇动离心管),然后快速移至冰浴中,静置2分钟;(4)向离心管中加入200μl的无菌的lb培养基(不加抗生素),混匀后置于摇床中37℃,220rpm,振摇1小时。目的是使质粒上相关的抗性标记基因表达,使菌体复苏;(5)涂布到相应抗性的固体培养基平皿中;(6)37℃培养箱中培养过夜。
64.6. 酶切验证,反应体系如表16:表16:验证体系
结果如图3和图4所示,其中图3为plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1(p3)电泳检测结果,图4为plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro(p4) 电泳检测结果,结果显示获得了目的质粒,分别为2132bp和2879bp大小的片段。
65.7. 测序挑取单菌落培养后提取质粒用sgfi/mlui双酶切进行酶切鉴定阳性克隆,并送测序验证。
66.实施例2:载体转染细胞,目的基因转录将构建的plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1(p3)载体和plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro(p4)载体共转染hek293细胞,puro筛选72小时的存活率验证及四个基因的mrna水平检测一、构建的plent
‑
ef1a
‑
oct4
‑
p2a
‑
sox2
‑
cmv
‑
n
‑
puro
‑
ts1载体和plent
‑
ef1a
‑
klf4
‑
p2a
‑
myc
‑
cmv
‑
ts2
‑
c
‑
puro载体共转染hek293细胞,puro筛选72小时的存活率验证1.1 正常生长的hek293细胞,铺6孔板;1.2 按照表17进行转染实验:1.2.1 细胞培养:取6孔板细胞培养皿,向每孔中加入2ml含1~2
×
105个细胞培养液,37℃ co2培养密度至40%~60%,密度过大,转染后不利筛选细胞。
67.1.2.2 转染液制备a液:用不含血清培养基稀释1μg质粒dna;b液:用不含血清培养基稀释3μl pei;分别将a液与b液轻弹混匀,静置5分钟。
68.1.2.3 吸取b液加入至a液中,轻弹混匀。室温中置25分钟。
69.1.2.4 把a/b复合物缓缓加入培养液中,摇匀,37℃温箱培养。
70.表17:转染体系1.3 转染48小时后进行拍照,曝光时间1s。结果如图5所示,均为转染结果:其中图5a为h2o+pei白光结果,图5b为h2o+pei 绿光显示结果,图5c为为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pei 白光结果,图5d为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pei 绿光结果,图5e为gfp
‑
puro+pei白光结果,图5f为gfp
‑
puro+pei绿光结果,结果显示转染过程正常,细胞均生长正常。
71.1.4 将细胞传代到10cm盘,同时加入嘌呤霉素,筛选72h,观察并拍照:1.5 实验结果:结果如图6所示,为筛选后细胞生长状态。其中,图6a为h2o+pei的白光结果,图6b为h2o+pei的绿光结果,图6c为ips
‑
n
‑
puro +ips
‑
c
‑
puro+pe1的白光结果,图6d为ips
‑
n
‑
puro+ips
‑
c
‑
puro+pe1的绿光结果,图6e为gfp
‑
puro+pei 白光结果,图6f为
gfp
‑
puro+pei绿光结果,结果显示经过嘌呤霉素筛选72h后,转染h2o+pei细胞组全部死亡;转染ips
‑
n
‑
puro+ips
‑
c
‑
puro+pe1质粒组的细胞存活率可达40%,与转染gfp
‑
puro质粒的对照组存活率基本一致。
72.1.6实验结论:分裂嘌呤霉素筛选标记的两个载体共转染获得了嘌呤霉素抗性。
73.二、ips
‑
n
‑
puro+ips
‑
c
‑
puro共转染,puro筛选后,ips相关的四个基因的mrna水平检测2.1收取以上转染实验的gfp
‑
puro+pei1组与ips
‑
n
‑
puro+ips
‑
c
‑
puro+pe12组细胞样本,提取rna,逆转录后进行mrna水平的检测。
74.2.2引物设计,如表18:表18:pcr引物2.3sybergreen法绝对定量pcr,qpcr上机时分别做3个复孔。
75.扩增体系见表19:表19:pcr扩增体系扩增条件:95℃3min,95℃5sec,60℃15sec,72℃15sec。循环数39。
76.2.4 实验结果见表20和图12(目的蛋白mrna在hek293细胞中表达拷贝数):表20:目的蛋白mrna表达拷贝数2.5 实验结论:使用分裂筛选标记的两个过表达载体,共同表达四个目的基因,这四个目的基因在mrna水平均得到明显过表达。
77.实施例3:包装慢病毒感染293a细胞筛选稳转细胞3.1 包装慢病毒产品:3.1.1 将hek293t细胞铺于10cm盘中,次日细胞融合度达到85%
‑
90%;3.1.2 按照比例配置转染试剂,混合后室温静置3min,后加入pei涡旋混匀,室温静置30min,加至10cm细胞培养皿中,转染后6小时更换新鲜培养基;3.1.3 观察转染效率并拍照;3.1.4 转染72小时后收取上清培养基,过0.45um滤膜;将过滤后的上清培养基加
入超滤管中进行超离,25000rpm,4℃,1.5hs;3.1.5 收集病毒。
78.3.2 检测病毒滴度:3.2.1 感染前6小时在24孔细胞板中以2.5
×
105细胞/孔密度接种hek293细胞;3.2.2 将慢病毒进行梯度稀释,共做三个梯度,然后加一组带荧光已知tu的慢病毒,混匀后加至铺好细胞的24孔板中;3.2.3 感染64
‑
68小时后收集细胞并进行基因组dna提取;3.2.4 qpcr检测ips
‑
n
‑
puro:plent
‑
ef1a
‑
oct4
‑
sox2
‑
cmv
‑
n
‑
puro
ꢀ‑
7.19e+08 iu/mlips
‑
c
‑
puro:plent
‑
ef1a
‑
klf4
‑
myc
‑
cmv
‑
c
‑
puro
ꢀ‑
1.87e+09 iu/ml3.3 感染方案:3.3.1 感染前6小时在6孔细胞板中以1.0
×
106细胞/孔密度接种hek293细胞;3.3.2 按照表19中分组,moi添加对应量的慢病毒。
79.分组见表21:表21:分组3.4 筛选稳转细胞:感染72小时后,加puro进行筛选,筛选一周可获得稳转细胞。结果如图8
‑
12所示,其中:图8为ips
‑
n
‑
puro
‑
moi 6+ips
‑
c
‑
puro
‑
moi 6组293a细胞的转染结果,图a和b分别为加压前和加压后的结果;结果显示该组细胞在经嘌呤霉素筛选后细胞呈稳定扩增状态,得到了稳转细胞系。
80.图9为ips
‑
n
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果;结果显示该组细胞不具有嘌呤霉素抗性,细胞全部死亡。
81.图10为ips
‑
c
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果;结果显示该组细胞不具有嘌呤霉素抗性,细胞全部死亡。
82.图11为lenti
‑
puro
‑
moi 12组293a细胞的转染结果,图a和b分别为加压前和加压后的结果;结果显示该组细胞不具有嘌呤霉素抗性,细胞全部死亡。
83.图12为空白对照组293a细胞的转染结果,图a和b分别为加压前和加压后的结果;结果显示该组细胞不具有嘌呤霉素抗性,细胞全部死亡。
84.3.5 定量检测ips稳转细胞系各基因在mrna表达量,结果如表22和图13(目的蛋白mrna在293细胞中的表达拷贝数)所示:表22:目的蛋白mrna表达拷贝数
以上结果显示,四个目的基因可以在293a细胞中稳定表达。
85.以上详细描述了本发明的优选实施方式,但是,本发明并不限于上述实施方式中的具体细节,在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,这些简单变型均属于本发明的保护范围。
86.另外需要说明的是,在上述具体实施方式中所描述的各个具体技术特征,在不矛盾的情况下,可以通过任何合适的方式进行组合,为了避免不必要的重复,本发明对各种可能的组合方式不再另行说明。
87.此外,本发明的各种不同的实施方式之间也可以进行任意组合,只要其不违背本发明的思想,其同样应当视为本发明所公开的内容。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1