三联吡啶铜配合物及其合成方法和应用
1.本发明涉及三联吡啶铜配合物及其合成方法和应用,属于医药技术领域。
背景技术:
2.三联吡啶及其衍生物在超分子化学、不对称催化、光敏及抗肿瘤等领域均有应用潜力。三联吡啶是由三个吡啶环组成的杂环化合物,特殊的结构使其能与金属形成稳定的配合物。近年来,三联吡啶配合物因具有良好的dna结合能力及抗肿瘤活性而被广泛研究。akhtar hussain等以三联吡啶衍生物为配体合成了一系列具有光活化抗肿瘤活性的镧系配合物(hussain a,gadadhar s,goswami t k,et al.photoactivated dna cleavage and anticancer activity of pyrenyl
‑
terpyridine lanthanide complexes[j].european journal of medicinal chemistry,2012,50(none):319
‑
331.)。milan m.等以三联吡啶衍生物为配体,合成了新型的单功能钌(ii)配合物,通过紫外
‑
可见光光度法、荧光猝灭法和粘度法等实验手段测定其dna结合能力,结果表明钌(ii)配合物通过插入dna碱基对之间的平面三联吡啶环与dna双螺旋相互作用,这些配合物对某些癌细胞有抑增殖作用(milutinovi
′
c m m,rilak a,bratsos i,et al.new 4
‑
(4
‑
chlorophenyl)
‑
2,2':6
′
,2
″‑
terpyridine ruthenium(ii)complexes:synthesis,characterization,interaction with dna/bsa and cytotoxicity studies[j].journal of inorganic biochemistry,2017,169:1
‑
12.)。
[0003]
三联吡啶铜(ii)配合物也已被设计与合成,这些铜(ii)配合物具有核酸酶活性、细胞毒性、较高的dna亲和力及抗肿瘤活性。biljana等合成了两个三联吡啶铜(ii)配合物,体外实验测定了配合物对人非小细胞肺癌(a549)具有良好的抗增殖性能,分子对接研究表明两个配合物均可与dna主沟相互作用,有较强的亲和力(biljanag,jasmina n
‑
r,tatjana i
‑
t,et al.synthesis,cytotoxic activity and dna
‑
binding properties of copper(ii)complexes with terpyridine[j].polyhedron,2018,139:313
‑
322.)。sofia gama等以三联吡啶和联吡啶为配体合成了几个新的铜(ii)配合物,这些配合物在没有外源性氧化剂或还原剂的情况下可以导致双链dna断裂,对卵巢癌a2780细胞具有较高的细胞毒性(是顺铂的的四倍)(gama s,rodrigues i,marques f,et al.new ternary bipyridine
–
terpyridine copper(ii)complexes as self
‑
activating chemical nucleases[j].rsc.advances,2014,4(106):61363
‑
61377.)。
技术实现要素:
[0004]
本发明要解决的技术问题是提供两例结构新颖的三联吡啶铜配合物及其合成方法和应用。
[0005]
为解决上述技术问题,本发明采用的技术方案如下:
[0006]
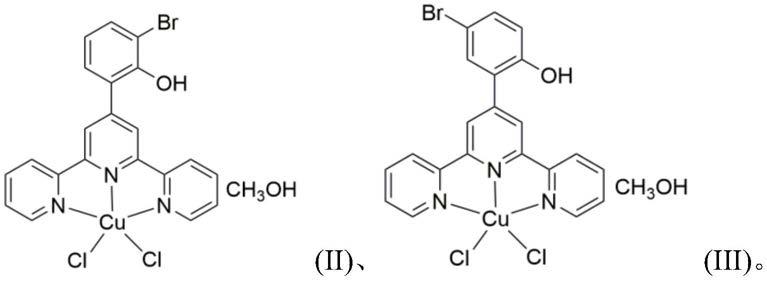
本发明所述的三联吡啶铜配合物为下述式(ii)或式(iii)所示化合物或其药学上可接受的盐:
[0007][0008]
本发明所述三联吡啶铜配合物的合成方法为取下述式(i)所示三联吡啶衍生物和二水合氯化铜溶于混合溶剂中,于加热条件下反应,反应物冷却,有晶体析出,收集晶体,即得目标产物;其中,
[0009]
当式(i)所示三联吡啶衍生物中r1=br、r2=h,且混合溶剂为由氯仿或二氯甲烷和甲醇按1:9.5~10.5的体积比组成的组合物时,得到的是式(ii)所示三联吡啶铜配合物;
[0010]
当式(i)所示三联吡啶衍生物中r1=h、r2=br,且混合溶剂为由氯仿或二氯甲烷和甲醇按1:3.5~4.5的体积比组成的组合物时,得到的是式(iii)所示三联吡啶铜配合物。
[0011][0012]
上述三联吡啶铜配合物的合成方法中,式(i)所示三联吡啶衍生物和二水合氯化铜的摩尔比为化学计量比,在实际的操作过程中,二水合氯化铜的用量可相对过量一些。
[0013]
上述三联吡啶铜配合物的合成方法中,反应优选在≥35℃的条件下进行,进一步优选在≥40℃的条件下进行,更优选在50~65℃的条件下进行。当反应在50~65℃条件下进行时,反应时间通常控制在24~72h。反应时通常采用一端封闭的厚壁硬质玻璃管来盛装料液。对于混合溶剂,其用量以能够溶解参加反应的原料为宜。具体地,以1mmol的式(i)所示三联吡啶衍生物为基准计算,全部原料所用混合溶剂的总用量通常为9~25ml,其中,当式(i)所示三联吡啶衍生物中r1=br、r2=h时,全部原料所用混合溶剂的总用量进一步优选为20~25ml;当式(i)所示三联吡啶衍生物中r1=h、r2=br时,全部原料所用混合溶剂的总用量进一步优选为9~11ml。
[0014]
上述式(i)所示三联吡啶衍生物可自行设计路线进行合成,优选按下述方法制备:
[0015]
取2
‑
乙酰基吡啶和3
‑
溴水杨醛或5
‑
溴水杨醛置于低碳醇中,加入氨水,之后调节体系的ph≥10,于加热或不加热条件下反应,所得反应物回收溶剂,收集固体,即得。
[0016]
在式(i)所示三联吡啶衍生物的制备方法中,所述的低碳醇为含1~4个碳原子的醇。具体为选自甲醇、乙醇、正丙醇和正丁醇中的一种或两种以上的组合,优选为甲醇和/或乙醇。所述低碳醇的用量以能够溶解参加反应的原料为宜。具体地,以1mmol的3
‑
溴水杨醛或5
‑
溴水杨醛为基准计算,全部原料所用低碳醇的总用量优选为2~3ml。
[0017]
在式(i)所示三联吡啶衍生物的制备方法中,反应通过薄层层析(tlc)跟踪检测,反应优选在加热条件下进行,进一步优选在35~50℃的条件下进行。当反应在35~50℃条件下进行时,反应时间通常控制在20~48h。反应完成后收集的固体可以进一步用合成时的溶剂洗涤以除去未反应的物质,优选采用甲醇和/或乙醇洗涤。
[0018]
在式(i)所示三联吡啶衍生物的制备方法中,只有在氨水存在的条件下才会有式(i)所示三联吡啶衍生物生成,所述氨水的加入量通常为3
‑
溴水杨醛或5
‑
溴水杨醛物质的量的1倍以上,优选为1倍。所述2
‑
乙酰基吡啶和3
‑
溴水杨醛或5
‑
溴水杨醛的摩尔比为化学计量比,在实际的操作过程中,2
‑
乙酰基吡啶的用量可相对过量一些。
[0019]
在式(i)所示三联吡啶衍生物的制备方法中采用现有技术中能使体系达到ph≥10条件的常规碱性物质调节体系的ph值,优选采用氢氧化钠、氢氧化钾、氢氧化钡等碱性物质来调节体系的ph值。进一步优选调节体系的ph≥12,更优选调节体系的ph=12~13。
[0020]
在本技术中,式(ii)所示三联吡啶铜配合物也称为配合物4,式(iii)所示三联吡啶铜配合物也称为配合物6;式(i)所示三联吡啶衍生物中,当r1=br、r2=h时的化合物也称为h
‑
l
c
或配体h
‑
l
c
,当r1=h、r2=br时的化合物也称为h
‑
l
d
或配体h
‑
l
d
。在具体合成时,当以2
‑
乙酰基吡啶和3
‑
溴水杨醛为原料时,对应得到的是配体h
‑
l
c
;当以2
‑
乙酰基吡啶和5
‑
溴水杨醛为原料时,对应得到的是配体h
‑
l
d
。
[0021]
申请人通过体外细胞生长抑制试验发现,上述三联吡啶铜配合物对多种肿瘤细胞具有极为显著的抑制细胞增殖效果,因此,本发明还包括上述三联吡啶铜配合物或其药学上可接受的盐在制备抗肿瘤药物中的应用。进一步的,本发明还包括一种药物组合物,含有治疗上有效剂量的上述三联吡啶铜配合物或其药效学上可接受的盐。
[0022]
与现有技术相比,本发明提供了两例结构新颖的三联吡啶铜配合物及其合成方法,申请人的试验结果表明,本发明所述化合物对多种肿瘤细胞株表现出显著的抑制作用,有望用作抗癌药物。
附图说明
[0023]
图1为配合物4的晶体结构(椭球率50%),省略溶剂分子。
[0024]
图2为配合物6的晶体结构(椭球率50%)。
[0025]
图3为配体h
‑
l
c
和配合物4的高效液相图,其中(a)为配体h
‑
l
c 0h,(b)为配体h
‑
l
c 48h,(c)为配合物4 0h,(d)为配合物4 48h。
[0026]
图4为配体h
‑
l
d
和配合物6的高效液相图,其中(a)为配体h
‑
l
d 0h,(b)为配体h
‑
l
d 48h,(c)为配合物6 0h,(d)为配合物6 48h。
[0027]
图5为空白对照及不同浓度的配合物4对bel
‑
7402作用48h后细胞周期的影响分布图。
[0028]
图6为不同浓度的配合物6对bel
‑
7402作用48h后细胞周期的影响分布图。
[0029]
图7为不同浓度的配合物4对bel
‑
7402细胞周期蛋白表达的影响图,其中(a)为不同浓度的配合物4对bel
‑
7402细胞不同蛋白的蛋白表达图,(b)为不同浓度的配合物4对bel
‑
7402细胞不同蛋白表达的柱状图,各蛋白柱状图中从左到右依次为空白对照、0.45μm、0.9μm、1.8μm。
[0030]
图8为不同浓度的配合物6对bel
‑
7402细胞周期蛋白表达的影响图,其中(a)为不
同浓度的配合物6对bel
‑
7402细胞不同蛋白的蛋白表达图,(b)为不同浓度的配合物6对bel
‑
7402细胞不同蛋白表达的柱状图,各蛋白柱状图中从左到右依次为空白对照、0.45μm、0.9μm、1.8μm。
[0031]
图9为不同浓度的配合物4对bel
‑
7402细胞凋亡的影响图。
[0032]
图10为不同浓度的配合物6对bel
‑
7402细胞凋亡的影响图。
[0033]
图11为不同浓度的配合物4对bel
‑
7402细胞线粒体膜电位的影响图。
[0034]
图12为不同浓度的配合物6对bel
‑
7402细胞线粒体膜电位的影响图。
[0035]
图13为不同浓度的配合物4对bel
‑
7402细胞内活性氧的影响图。
[0036]
图14为不同浓度的配合物6对bel
‑
7402细胞内活性氧的影响图。
[0037]
图15为不同浓度的配合物4和6对bel
‑
7402细胞中ca
2+
浓度的影响图,其中(a)为配合物4(未标注的那条曲线为control),(b)为配合物6(未标注的那条曲线为control)。
[0038]
图16为不同浓度的配合物4对bel
‑
7402细胞中caspase
‑
3/9表达水平的影响。
[0039]
图17为不同浓度的配合物6对bel
‑
7402细胞中caspase
‑
3/9表达水平的影响。
[0040]
图18为配合物4对bel
‑
7402细胞凋亡相关蛋白表达量的影响图,其中(a)为不同浓度的配合物4对bel
‑
7402细胞凋亡相关蛋白的蛋白表达图,(b)为不同浓度的配合物4对bel
‑
7402细胞凋亡相关蛋白表达量的柱状图,各蛋白柱状图中从左到右依次为空白对照、0.45μm、0.9μm、1.8μm。
[0041]
图19为配合物6对bel
‑
7402细胞凋亡相关蛋白表达量的影响图,其中(a)为不同浓度的配合物6对bel
‑
7402细胞凋亡相关蛋白的蛋白表达图,(b)为不同浓度的配合物6对bel
‑
7402细胞凋亡相关蛋白表达量的柱状图,各蛋白柱状图中从左到右依次为空白对照、0.45μm、0.9μm、1.8μm。
具体实施方式
[0042]
为了更好的解释本发明的技术方案,下面结合实施例及附图对本发明作进一步详细的描述,但本发明的实施方式不限于此。若未特别指明,实施例中所用的技术特征可以替换为具有在不背离发明构思前提下等同或相似功能或效果的其他本领域已知的技术特征。
[0043]
实施例1:配体h
‑
l
c
的制备
[0044]
在100ml圆底烧瓶中加入2
‑
乙酰基吡啶(2.24ml,20mmol)、3
‑
溴水杨醛(2.01g,10mmol),量取50ml乙醇加入到圆底烧瓶中,摇匀,再量取0.75ml浓度为25%的氨水(加入的氨水约为10mmol)加入其中,称取氢氧化钾0.14g小心加入到圆底烧瓶中(氢氧化钾溶解搅拌均匀后体系的ph值在12~13之间),放入磁子,安装好回流反应装置后,在40℃水浴下反应24小时(起初溶液为淡褐色,随着反应的进行有浅黄色固体产生)。反应结束后,将圆底烧瓶取下,进行旋蒸,将瓶内溶剂蒸出一半,之后进行抽滤,收集滤饼,用乙醇洗涤,干燥,得浅黄色粉末状固体。产率为92.3%(3.72g,基于3
‑
溴水杨醛为基准计算)。
[0045]
对本实施例所得产物进行表征:
[0046]
(1)高分辨质谱:esi
–
ms m/z:405.4[m+h]
+
,其中m为配体h
‑
l
a
的分子量。
[0047]
(2)核磁氢谱:1h nmr(500mhz,dmso
‑
d6):
[0048]
δ15.71(s,1h),9.08(d,j=1.3hz,1h),8.90(d,j=1.2hz,1h),8.86
–
8.84(m,2h),8.49(d,j=8.2hz,1h),8.37(d,j=6.9hz,1h),8.23(d,j=7.9hz,1h),8.13(td,j=7.7,
1.7hz,1h),8.09
–
8.05(m,1h),7.72
–
7.69(m,1h),7.61
–
7.57(m,2h),6.96(d,j=7.9hz,1h)。
[0049][0050]
(3)核磁碳谱:
13
c nmr(125mhz,dmso
‑
d6):
[0051]
δ156.98,156.41,153.60,153.52,153.14,150.52,150.50,149.71,138.49,138.20,135.27,127.50,125.49,125.47,122.52,121.12,120.42,120.33,117.75,117.57,111.92.
[0052]
(4)元素分析(c
21
h
14
brn3o):
[0053]
计算值(%):c62.39,h3.49,n10.39。
[0054]
实测值(%):c 62.36,h 3.55,n10.37。
[0055]
(5)红外光谱:selected ir(kbr,cm
–1):
[0056]
3491,3054,3013,1563,1470,1392,1226,1177,1078,795。
[0057]
由此,可以确定本实施例所得浅黄色粉末状固体为目标产物配体h
‑
l
c
。
[0058]
实施例2:配体h
‑
l
c
的制备
[0059]
重复实施例1,不同的是,用甲醇代替乙醇,用氢氧化钠代替氢氧化钾,控制氢氧化钠的加入量为调节体系的ph=12,反应在50℃水浴下进行。结果得到浅黄色粉末状固体。产率为90.6%(基于基于3
‑
溴水杨醛为基准计算)。
[0060]
对本实施例所得产物进行高分辨质谱分析、核磁表征、元素分析及红外分析,确定所得的浅黄色粉末状固体为目标产物配体h
‑
l
c
。
[0061]
实施例3:配体h
‑
l
d
的制备
[0062]
重复实施例1,不同的是,用5
‑
溴水杨醛代替3
‑
溴水杨醛。结果得到浅黄色粉末状固体3.67g。产率为91.1%(基于5
‑
溴水杨醛为基准计算)。
[0063]
对本实施例所得产物进行表征:
[0064]
(1)高分辨质谱:esi
–
ms m/z:404.04[m+h]
+
,其中m为配体h
‑
l
d
的分子量。
[0065]
(2)核磁氢谱:1h nmr(400mhz,dmso
‑
d6):
[0066]
δ10.31(s,1h),8.74(ddd,j=4.8,1.8,0.9hz,2h),8.68(t,j=1.1hz,1h),8.66(d,j=1.1hz,1h),8.65(s,2h),8.03(td,j=7.7,1.8hz,2h),7.65(d,j=2.5hz,1h),7.54
–
7.51(m,2h),7.50(dd,j=3.3,1.8hz,1h),7.03(dd,j=8.7,3.0hz,1h)。
[0067]
(3)核磁碳谱:
13
c nmr(101mhz,dmso
‑
d6):
[0068]
δ155.55,155.39,154.71,154.54,149.81,147.25,147.22,137.91,133.23,132.51,127.85,124.90,121.32,121.12,119.11,111.29,111.23。
[0069]
(4)元素分析(c
21
h
14
brn3o):
[0070]
计算值(%):c62.39,h3.49,n10.39。
[0071]
实测值(%):c62.37,h 3.57,n10.34。
[0072]
(5)红外光谱:selectedir(kbr,cm
–1):
[0073]
3545,3049,3012,1585,1567,1471,1385,1282,1256,1239,790。
[0074]
由此,可以确定本实施例所得浅黄色粉末状固体为目标产物配体h
‑
l
d
。
[0075]
实施例4:配体h
‑
l
d
的制备
[0076]
重复实施例3,不同的是,用正丙醇代替乙醇,氨水的加入量改为2ml,控制氢氧化钾的加入量为调节体系的ph=13,反应在35℃水浴下进行,反应时间改为48小时。结果得到浅黄色粉末状固体。产率为84.3%(基于5
‑
溴水杨醛为基准计算)。
[0077]
对本实施例所得产物进行高分辨质谱分析、核磁表征、元素分析及红外分析,确定所得的浅黄色粉末状固体为目标产物配体h
‑
l
d
。
[0078]
实施例5:配体h
‑
l
d
的制备
[0079]
重复实施例3,不同的是,用正丁醇代替乙醇,反应在25℃室温下进行,反应时间改为72小时。结果得到浅黄色粉末状固体。产率为80.2%(基于5
‑
溴水杨醛为基准计算)。
[0080]
对本实施例所得产物进行高分辨质谱分析、核磁表征、元素分析及红外分析,确定所得的浅黄色粉末状固体为目标产物配体h
‑
l
d
。
[0081]
实施例6:式(ii)所示三联吡啶铜配合物[cu(h
‑
l
c
)cl2]
·
ch3oh(即配合物4)的制备
[0082]
称取10mg(0.025mmol)配体h
‑
l
c
和10mg(0.056mmol)cucl2.2h2o于一端闭合的25cm pyrex厚壁玻璃管中,滴加0.05ml氯仿与0.5ml甲醇(氯仿和甲醇的体积比为1:10),将玻璃管熔封后置于65℃的烘箱中反应72小时。反应结束后梯度降温至室温,可观察到管内有绿色块状晶体生成,收集晶体,干燥。产率为78.3%(11.08mg,基于配体h
‑
l
c
为基准计算)。
[0083]
挑选出大小、形状适合的单晶,采用单晶x
‑
射线衍射分析等方法确定其结构,具体表征数据如下:
[0084]
(1)高分辨质谱:esi
–
ms m/z:503.0[m
‑
ch3oh
‑
cl]
+
,其中m为配合物4的分子量。
[0085]
(2)元素分析(c
21
h
14
brcl2cun3o):
[0086]
计算值(%):c 46.82,h 2.62,n 7.80。
[0087]
实测值(%):c 46.77,h 2.75,n 7.76.
[0088]
(3)红外分析selectedir(kbr,cm
–1):
[0089]
ν(o
‑
h)3411(s);ν(ar
‑
h)3048(m);ν(c
‑
o)1230(s);ν(c
‑
br)782(s)。
[0090]
(4)单晶x
‑
射线衍射分析:
[0091]
选取尺寸适中的绿色块状晶体置于安捷伦supernova x
‑
射线单晶衍射仪上,采用经石墨单色器单色化的射线作为光源进行单晶结构测定。本实施例所得产物的初始晶体结构均采用shelxs
‑
97直接法解出,衍射数据经lp因子和经验吸收校正,对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法精修,所有氢原子坐标由理论计算确定。所得晶体学和结构精修数据如下述表1所示,部分键长键角数据分别如下述表2所示,所得绿色块状晶体的晶体结构如图1所示,平面结构如下述式(ii)所示。因此,确定所得的绿色块状晶体为本发明目标产物式(ii)所示三联吡啶铜配合物[cu(h
‑
l
c
)cl2]
·
ch3oh,即配合物4。
[0092][0093]
表1配合物4和6的晶体学和结构修正数据
[0094][0095]
a
r1=σ||f
o
|
–
|f
c
||/σ|f
o
|;
b
ωr2=[σw(f
o2
–
f
c2
)2/σw(f
o2
)2]
0.5
.
[0096]
表2配合物4和6的部分键长和键角[
°
]
[0097][0098][0099]
单晶x
‑
射线衍射分析结果表明:配合物4属于单斜晶系,p21/c空间群,中心cu(ⅱ)离子与两个氯离子以及来自配体h
‑
l
c
的三个n原子配位,形成五配位扭曲四方锥几何构型。
[0100]
对比例6
[0101]
①
重复实施例6,不同的是,滴加0.05ml氯仿和0.55ml甲醇(氯仿和甲醇的体积比为1:11)。结果没有目标产物的晶体或沉淀生成。
[0102]
②
重复实施例6,不同的是,滴加0.05ml氯仿和0.45ml甲醇(氯仿和甲醇的体积比为1:9)。结果没有目标产物的晶体或沉淀生成。
[0103]
③
重复实施例6,不同的是,将混合溶剂改为以单一的甲醇为溶剂。结果没有目标产物的晶体或沉淀生成。
[0104]
④
重复实施例6,不同的是,将混合溶剂中的氯仿用丙酮、乙腈、dmf或dmso等替代。结果均没有目标产物的晶体或沉淀生成。
[0105]
⑤
重复实施例6,不同的是,用cuso4.5h2o或cu(clo4)2.6h2o代替cucl2.2h2o,希望得到本发明目标产物,但均未得到晶体或沉淀,说明用cuso4.5h2o或cu(clo4)2.6h2o无法达到形成本发明所述铜配合物和结晶的热力学条件。
[0106]
实施例7:配合物4的制备
[0107]
重复实施例6,不同的是,滴加0.05ml二氯甲烷和0.525ml甲醇(二氯甲烷和甲醇的体积比为1:10.5),反应改在40℃水浴下进行,反应时间改为96小时。结果得到绿色块状晶体。产率65.2%(9.23mg,基于配体h
‑
l
c
为基准计算)。
[0108]
对本实施例所得产物进行高分辨质谱分析、元素分析、红外分析及单晶衍射分析,确定所得的绿色块状晶体为本发明目标产物配合物4。
[0109]
实施例8:配合物4的制备
[0110]
重复实施例6,不同的是,滴加0.05ml氯仿和0.48ml甲醇(氯仿和甲醇的体积比为1:9.6)。结果得到绿色块状晶体。产率74.7%(10.6mg,基于配体h
‑
l
c
为基准计算)。
[0111]
对本实施例所得产物进行高分辨质谱分析、元素分析、红外分析及单晶衍射分析,
确定所得的绿色块状晶体为本发明目标产物配合物4。
[0112]
实施例9:配合物4的制备
[0113]
重复实施例6,不同的是,反应改在50℃水浴下进行。结果得到绿色块状晶体。产率72.3%(10.2mg,基于配体h
‑
l
c
为基准计算)。
[0114]
对本实施例所得产物进行高分辨质谱分析、元素分析、红外分析及单晶衍射分析,确定所得的绿色块状晶体为本发明目标产物配合物4。
[0115]
实施例10:式(iii)所示三联吡啶铜配合物[cu(h
‑
l
d
)cl2]
·
ch3oh(即配合物6)的制备
[0116]
称取10mg(0.025mmol)配体h
‑
l
d
和10mg(0.056mmol)cucl2.2h2o于一端闭合的25cm pyrex厚壁玻璃管中,滴加0.05ml氯仿与0.2ml甲醇(氯仿和甲醇的体积比为1:4),将玻璃管开口端熔封后置于65℃的烘箱中反应72小时。反应结束后梯度降温至室温,可观察到管内有蓝色块状晶体生成,收集晶体,干燥。产率为74.2%(10.5mg,基于配体h
‑
l
d
为基准计算)。
[0117]
挑选出大小、形状适合的单晶,采用单晶x
‑
射线衍射分析等方法确定其结构,具体表征数据如下:
[0118]
(1)高分辨质谱:esi
–
ms m/z:502.9[m
‑
ch3oh
‑
cl]
+
,其中m为配合物6的分子量。
[0119]
(2)元素分析(c
21
h
14
brcl2cun3o):
[0120]
计算值(%):c 46.82,h 2.62,n 7.80。
[0121]
实测值(%):c 46.68,h 2.71,n 7.76。
[0122]
(3)红外分析selectedir(kbr,cm
–1):
[0123]
ν(o
‑
h)3376(s);ν(ar
‑
h)3065(s);ν(c
‑
o)1286(s);ν(c
‑
br)785(s)。
[0124]
(4)单晶x
‑
射线衍射分析:
[0125]
选取尺寸适中的蓝色块状晶体置于安捷伦supernova x
‑
射线单晶衍射仪上,采用经石墨单色器单色化的射线作为光源进行单晶结构测定。本实施例所得产物的初始晶体结构均采用shelxs
‑
97直接法解出,衍射数据经lp因子和经验吸收校正,对全部非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法精修,所有氢原子坐标由理论计算确定。所得晶体学和结构精修数据如上述表1所示,部分键长键角数据分别如上述表2所示,所得蓝色块状晶体的晶体结构如图2所示,平面结构如下述式(iii)所示。因此,确定所得的蓝色块状晶体为本发明目标产物式(iii)所示三联吡啶铜配合物[cu(h
‑
l
d
)cl2]
·
ch3oh,即配合物6。
[0126][0127]
单晶x
‑
射线衍射分析结果表明:配合物6属于三斜晶系,pˉ1空间群,中心铜(ⅱ)离子与两个氯离子以及来自配体h
‑
l
d
的三个n原子配位,形成五配位扭曲四方锥几何构型。
[0128]
对比例10
[0129]
①
重复实施例10,不同的是,滴加0.05ml氯仿和0.25ml甲醇(氯仿和甲醇的体积比为1:5)。结果没有目标产物的晶体或沉淀生成。
[0130]
②
重复实施例10,不同的是,滴加0.05ml氯仿和0.15ml甲醇(氯仿和甲醇的体积比为1:3)。结果没有目标产物的晶体或沉淀生成。
[0131]
③
重复实施例10,不同的是,将混合溶剂改为以单一的甲醇为溶剂。结果没有目标产物的晶体或沉淀生成。
[0132]
④
重复实施例10,不同的是,将混合溶剂中的氯仿用丙酮、乙腈、dmf或dmso等替代。结果均没有目标产物的晶体或沉淀生成。
[0133]
⑤
重复实施例10,不同的是,用cuso4.5h2o或cu(clo4)2.6h2o代替cucl2.2h2o,希望得到本发明目标产物,但均未得到晶体或沉淀,说明用cuso4.5h2o或cu(clo4)2.6h2o无法达到形成本发明所述铜配合物和结晶的热力学条件。
[0134]
实施例11:配合物6的制备
[0135]
重复实施例10,不同的是,滴加0.05ml氯仿和0.225ml甲醇(氯仿和甲醇的体积比为1:4.5),反应时间改为48小时。结果得到蓝色块状晶体。产率71.5%(10.1mg,基于配体h
‑
l
d
为基准计算)。
[0136]
对本实施例所得产物进行高分辨质谱分析、元素分析、红外分析及单晶衍射分析,确定所得的蓝色块状晶体为本发明目标产物配合物6。
[0137]
实施例12:配合物6的制备
[0138]
重复实施例10,不同的是,滴加0.05ml二氯甲烷和1.75ml甲醇(二氯甲烷和甲醇的体积比为1:3.5),反应改在35℃水浴下进行,反应时间改为96小时。结果得到蓝色块状晶体。产率58.6%(8.3mg,基于配体h
‑
l
d
为基准计算)。
[0139]
对本实施例所得产物进行高分辨质谱分析、元素分析、红外分析及单晶衍射分析,确定所得的蓝色块状晶体为本发明目标产物配合物6。
[0140]
实验例1:本发明所述配体及配合物稳定性实验
[0141]
1.1紫外
‑
可见光谱法测定化合物稳定性
[0142]
申请人应用紫外
‑
可见光谱法来检测所合成化合物在ph 7.35的tris
‑
hcl缓冲液中的稳定性,为后续的细胞水平实验提供实验基础。
[0143]
将浓度为2
×
10
‑5mol/l的配体及配合物在ph 7.35的tris
‑
hcl缓冲液中,置于室温下0和48小时后,分别用紫外光谱仪各测一次吸光度。实验结果显示,各组吸收峰没有发生红移或蓝移现象,也没有新的吸收峰出现,说明本发明所合成的配体及配合物在tris
‑
hcl缓冲液中能够稳定存在至少48h。
[0144]
1.2高效液相色谱法(hplc)测定化合物稳定性及纯度
[0145]
图3和图4分别是配体h
‑
l
c
及其配合物4、h
‑
l
d
及其配合物6的高效液相色谱图(所有高效液相色谱图(lc
‑
20at,spd
‑
20ahplccolumn,150mm
×
5.0μm i.d.,株温:40℃,流动相:methol/h2o(90:10),流速:1.0ml/min,进样量:1.50
×
10
‑4m))。液相色谱实验表明:48小时后配体及配合物没有新的峰出现,而且峰的位移几乎没有变化,说明在室温下配体及配合物在dmso储备液中能稳定存在至少48小时,实验结果与用紫外
‑
可见光谱法测定结果一致,说明化合物具备足够的稳定性。此外,hplc实验也说明了化合物纯度皆在99%以上,可以进行细胞实验。
[0146]
实验例2:本发明所述配体及配合物体外抗肿瘤活性研究
[0147]
2.1实验仪器及试剂,如下述表3所示。
[0148]
表3实验所用仪器与试剂
[0149][0150]
2.2细胞株及培养
[0151]
本例所用细胞株有:mgc80
‑
3胃癌细胞、t
‑
24人膀胱癌细胞、nci
‑
h460人肺癌细胞、hela人宫颈癌细胞、bel
‑
7402人肝癌细胞、hep
‑
g2肝癌细胞、sk
‑
ov
‑
3卵巢癌细胞以及hl
‑
7702人正常肝细胞。所有细胞株均在5%co2、37℃的培养箱中进行培养。使用倒置显微镜观察细胞生长状态,待细胞贴壁80~90%后用pbs缓冲液洗涤细胞2次,再用胰蛋白酶消化,取对数生长期的细胞进行实验。
[0152]
2.3mtt法测定化合物细胞水平的ic
50
值
[0153]
①
消化细胞,取状态良好的对数生长期细胞于96孔培养板中,每孔180μl(约4500
‑
5000个细胞);
[0154]
②
待细胞贴壁生长至约2/3的孔面积后,每孔加入20μl的样品,平行设5个复孔,同时设置相应的空白对照组;
[0155]
③
培养48h后,每孔加入10μl mtt试剂(浓度为5mg/mlpbs);
[0156]
④
孵育4~6h后,将培养液吸弃,每孔加入150μl dmso,震荡5~8min,使甲瓒结晶充分溶解;
[0157]
⑤
使用酶标仪测定od值(波长570/630nm)。
[0158]
对筛选化合物,通常设置5个浓度梯度(20,10,5,2.5,1.25μm),但对于活性较好的配合物,设置的浓度梯度为(6,3,1.5,0.75,0.375μm),所有实验均重复3次以上取平均值。对本发明合成的化合物均使用分析纯的dmso来溶解,顺铂则使用生理盐水来溶解。
[0159]
2.4结果与讨论
[0160]
本实验利用bliss法计算得到了本发明合成的化合物对7种肿瘤细胞株和1种正常细胞株的ic
50
值(如表4所示,单位:μm)。
[0161]
表4配体h
‑
l
c
和h
‑
l
d
及配合物4和6对不同细胞株的ic
50
值(μm)
[0162][0163]
由表4可知,配体h
‑
l
c
和h
‑
l
d
和配合物4和6对所选的肿瘤细胞株的抑制作用均高于顺铂,h
‑
l
c
和h
‑
l
d
对所选肿瘤细胞株的ic
50
值都在7μm内,而配合物4和6对所选肿瘤细胞株的ic
50
值都在3μm内。其中,配合物4和6对bel
‑
7402人肝癌细胞的抑制作用最高,ic
50
值分别为0.83
±
0.12、和0.74
±
0.07μm,远低于顺铂的ic
50
值(17.10
±
0.87μm)。同时,由表4可知配合物4和6对bel
‑
7402人肝癌细胞的选择性抑制指数分别为2.7、3.8(选择性抑制指数=正常细胞的ic
50
/7402的ic
50
,即配合物4的选择性抑制指数=2.25/0.83=2.7,配合物6的选择性抑制指数=2.79/0.74=3.8)。因配合物对bel
‑
7402人肝癌细胞在一定程度上具有选择性抑制作用,所以对配合物4和6进行初步的抗肿瘤机制研究。
[0164]
实验例3:本发明所述配合物体外抗肿瘤作用机制研究
[0165]
3.1实验仪器与试剂,如下述表5所示。
[0166]
表5实验所用仪器及试剂
[0167]
[0168][0169]
3.2实验方法
[0170]
3.2.1流式细胞术检测细胞周期
[0171]
在肿瘤细胞中,细胞周期与细胞凋亡密切相关。碘化丙啶(pi)可以透过受损的细胞膜,对凋亡细胞的细胞核进行染色,根据荧光的强弱程度来分析细胞内的dna含量,从而检测细胞周期阻滞的情况。
[0172]
①
消化生长状态良好的对数期细胞,取约2
×
106个细胞接种于70mm的培养皿中,置于培养箱。
[0173]
②
待细胞贴壁至约2/3皿面积后,加入配置好的样品与细胞共培养48小时,设置空白对照组。
[0174]
③
用pbs洗涤细胞2~3次,胰蛋白酶消化并将所有细胞收集与15ml离心管中。
[0175]
④
1000rpm/min离心10分钟,倒掉上清液,加入pbs洗涤后再离心。
[0176]
⑤
吸除pbs,加入1mlpbs吹匀细胞后,边震荡边加入9ml冷冻后的消毒酒精,放入
‑
20℃过夜。
[0177]
⑥
取出后,1500rpm/min离心10分钟,弃去上清液后再用pbs洗涤。
[0178]
⑦
加入0.5ml rnase a(50μg/ml),吹匀细胞后,37℃孵育20~30min。
[0179]
⑧
加入20μl pi(50μg/ml),避光染色后过膜,用流式细胞仪检测细胞周期。
[0180]
3.2.2吸收和分布实验
[0181]
icp
‑
ms(电感耦合等离子体质谱法)是一种灵敏度高,可以快速检测细胞内金属含量的方法[17]。本节采用icp
‑
ms检测了配合物4、6在bel
‑
7402细胞中的摄取和分布,实验步骤如下:
[0182]
①
培养、加药及收集细胞的方法参考4.2.1的步骤
①
、
②
和
③
。
[0183]
②
按细胞线粒体与细胞浆蛋白抽提试剂盒和细胞核蛋白与细胞浆蛋白抽提试剂盒说明书提取细胞中不同的细胞器。
[0184]
③
二次水定容至1ml后,按顺序加入1ml浓hno3和2.5ml 30%h2o2,加二次水至总体积为10ml,过夜。
[0185]
④
取1ml,再次加入二次水定容至10ml,过膜后上机测试。
[0186]
3.2.3蛋白质印迹法(western blot)法检测配合物对周期相关蛋白表达的影响
[0187]
蛋白质印迹法常常被用来检测蛋白质表达水平,基本实验步骤如下:
[0188]
(1)提取蛋白
[0189]
①
培养、加药及收集细胞的方法参考3.2.1的步骤
①
、
②
和
③
。
[0190]
②
加入适量的裂解液(含pmsf,浓度为1mm)后转移至1.5ml离心管中,置于冰上裂解30min。
[0191]
③
4℃离心15min(13000rpm),取上清液并分装少量于新的离心管中用于测定蛋白浓度。
[0192]
④
按体积比3:1(上清液:4*sds
‑
page蛋白上样缓冲液)加入上样缓冲液,混匀后煮沸。
[0193]
⑤
自然冷却后置于
‑
20℃保存。
[0194]
(2)蛋白定量
[0195]
按bca蛋白定量试剂盒处理蛋白样品,在96孔板中加入各种试剂(如表6所示),37℃孵育30分钟后,用酶标仪测定在波长562nm处的吸光度值,绘制标准曲线并计算样品总蛋白量的相对浓度。
[0196]
表6 bca标准曲线制作表
[0197][0198]
(3)sds
‑
page电泳
[0199]
表7分离胶与浓缩胶配制方法
[0200]
[0201][0202]
①
制胶:按照表7配制分离胶与浓缩胶,用移液枪移取适量体积的分离胶加入夹好的玻璃板中,加入二次水覆盖。待凝固后倒掉上层的水。将浓缩胶灌满玻璃板后插入梳子,凝固后将梳子拔出。
[0203]
②
上样:把制好的胶放入电泳槽,加电泳液,按顺序上样(样品20μg/lane,marker 4μl)。
[0204]
③
电泳:80v、150min,待溴酚蓝即将跑出时停止。
[0205]
④
转膜:剪好pvdf膜,在无水甲醇中活化2min。按海绵垫、滤纸、凝胶、pvdf膜、滤纸、海绵垫的顺序放置,放入转膜槽中,再加入预冷的转膜液。pvdf膜在正极,凝胶在负极,电流250a,时间按照目的蛋白的分子量进行调整。
[0206]
⑤
免疫反应:tbst洗pvdf膜两次,每次5min,转移至封闭液中2h。用tbst洗三次封闭后的膜,用滤纸吸干tbst后将膜放入稀释后的一抗里面,4℃孵育过夜。tbst洗三次后将膜放入稀释后的二抗中室温孵育1h。tbst洗膜三次,每次10min。
[0207]
⑥
化学发光:化学发光液试剂a与试剂b等体积混合,吸取适量的混合液与膜充分接触,曝光1~10min。
[0208]
⑦
凝胶成像与图像分析。
[0209]
3.2.4流式细胞术检测细胞凋亡
[0210]
本实验采用annexin
‑
v
‑
fitc和pi对细胞进行双重染色,通过流式细胞术定量地检测细胞发生凋亡的百分数。实验操作步骤如下:
[0211]
①
消化对数期细胞,接种于六孔板(每孔2ml,约1
×
106个细胞);
[0212]
②
待细胞贴壁生长至孔面积的80~90%时,加入化合物与细胞共培养48h。
[0213]
③
收集细胞,pbs洗涤(2次),离心5min(1200rpm/min),弃去上清液。
[0214]
④
按凋亡试剂盒说明书对细胞进行染色,应用流式细胞仪检测细胞凋亡。
[0215]
3.2.5线粒体膜电位(δψm)的检测
[0216]
流式细胞术可以检测δψm的变化,从而分析细胞凋亡的情况。实验过程:
[0217]
①
培养细胞、加药孵育及收集细胞参考3.2.1的第
①
、
②
、
③
步。
[0218]
②
按照线粒体膜电位检测试剂盒(jc
‑
1)说明书对细胞进行染色,应用流式细胞仪检测δψm的变化。
[0219]
3.2.6细胞内活性氧(ros)检测
[0220]
dcfh
‑
da荧光探针可检测细胞内活性氧水平的变化,其本身无荧光,但可以被ros氧化为有荧光的dcf,通过检测细胞内的dcf即可了解细胞内ros水平的变化。具体实验步骤如下:
[0221]
①
培养细胞及加药的方法参考3.2.1的步骤
①
、
②
。
[0222]
②
用无血清的培养基洗细胞2~3遍,往六孔板中加入稀释好的dcfh
‑
da,每孔1ml,避光染色20min。
[0223]
③
用pbs洗涤细胞2~3遍,再加入1ml无血清的培养基,使用荧光倒置显微镜检测细胞内活性氧水平的变化。
[0224]
3.2.7细胞内ca
2+
浓度检测
[0225]
使用流式细胞仪可以检测细胞内钙离子水平的变化,ex/em=506nm/526nm。操作如下:
[0226]
①
培养细胞、加药孵育及收集细胞参考3.2.1的第
①
、
②
、
③
步。
[0227]
②
按钙离子检验试剂盒说明书对细胞进行染色,应用流式细胞仪检测ca2+浓度。
[0228]
3.2.8 caspase
‑
3/9的活性检测
[0229]
采用caspglowtm fluorescein active caspase
‑
3/9staining kit试剂盒来检测caspase
‑
3/9。简单操作过程如下:
[0230]
①
培养细胞、加药孵育及收集细胞参考4.2.1的第
①
、
②
、
③
步。
[0231]
②
用300μl pbs重悬细胞,加入1μl的caspase
‑
3/9染液,混匀后37℃避光孵育30min。
[0232]
③
离心3min(1200rpm/min),除去工作液,用相应的caspase
‑
3/9buffer洗涤2~3次。
[0233]
④
离心后重新加入0.5ml的buffer,过膜后用流式细胞仪检测。
[0234]
3.2.9western blot检测配合物对凋亡相关蛋白表达的影响
[0235]
实验步骤参考4.2.2。
[0236]
3.3结果与讨论
[0237]
3.3.1流式细胞术检测细胞周期
[0238]
细胞周期是一个参与细胞生长、组织再生、dna修复和细胞凋亡等事件的高度有序的过程。真核细胞的细胞周期分为两个主要阶段:间期和分裂期,其中,间期主要由由三个阶段组成:g1期(dna合成前期),s期(dna合成期),g2期(胞有丝分裂准备期)。细胞周期调控需要大量信号的参与,信号缺失则会导致细胞周期阻滞。当癌细胞在外界条件影响下发生周期阻滞时,细胞增殖被抑制,同时也有可能诱导细胞凋亡。
[0239]
图4.2是bel
‑
7402细胞与配合物4(0.4、0.8和1.6μm)孵育48h后的细胞周期分布图。与对照组相比,配合物4对bel
‑
7402细胞周期有较大干扰,g1期的细胞由48.92%增至66.57%、71.25%、74.38%,相对含量分别增加了:17.65%、22.33%、25.46%。
[0240]
图4.4是bel
‑
7402细胞与配合物6(0.35、0.7和1.4μm)孵育48h后的细胞周期分布图。与对照组相比,配合物6作用于bel
‑
7402细胞后,g1期的细胞由51.33%增至74.53%、74.72%、75.23%,g1期的细胞相对含量分别增加了:23.20%、23.39%、23.90%。
[0241]
3.3.2配合物4、6在bel
‑
7402细胞中的吸收与分布
[0242]
电感耦合等离子体
‑
质谱法(icp
‑
ms)是种可以测定痕量元素的无机质谱技术,其广泛应用于地质、环境、生物及医药行业。为了进一步了解药物的抗肿瘤机制,本节应用icp
‑
ms检测药物在细胞内的积累情况。
[0243]
表8和9分别是配合物4(0.8μm)、配合物6(0.7μm)作用bel
‑
7402细胞48小时后在细胞内各细胞器的分布情况。
[0244]
表8配合物4中cu在bel
‑
7402细胞内各细胞器的分布情况
[0245][0246]
表9配合物6中cu在bel
‑
7402细胞内各细胞器的分布情况
[0247][0248]
结果显示:相比对照组,配合物4,6作用细胞48h后,cu在细胞膜、细胞核及线粒体内的含量均有增加,且在线粒体内的增量较多。配合物4作用后,cu在线粒体的含量由1.567μg/106cells增至9.280μg/106cells,约为空白组的6倍,而配合物6作用后,cu在线粒体的含量由1.567μg/106cells增至5.950μg/106cells,约为空白组的4倍。这说明配合物4,6可以进入bel
‑
7402细胞的线粒体中,而且可以在线粒体内有效积累。
[0249]
3.3.3western blot检测配合物对肿瘤细胞周期相关蛋白的影响
[0250]
为确保细胞周期有序进行,大量的蛋白质参与了细胞周期的调控,细胞周期检查点是保证细胞周期准确无误进行的检查机制。检查点传感器chk1(checkpoint kinase 1)和chk2(checkpoint kinase 2)属于丝氨酸/苏氨酸蛋白激酶家族,也是顶端检查点激酶atm和atr的底物。哺乳动物细胞对dna损伤的主要检查点反应是atm(atp)/chk2(chk1)
‑
p53
‑
p21通路,它可以诱导长时间甚至是永久性的g1期阻滞。dna复制检查点(即s期内阶段检查点)在dna复制异常时被激活,此时效应激酶chk1使下游的cdc25a磷酸化,cdc25a主要在g1
‑
s期过渡期间激活cyclin e
‑
cdk2和cyclin a
‑
cdk2复合物。所以,细胞可以通过chk1/cdc25a
‑
cdk2通路来阻止dna复制,并将细胞阻滞于s期,而当cyclin e/a
‑
cdk2复合物失活时,也会导致细胞被阻滞在g1期。
[0251]
图7和8分别为western blot检测配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞chk1、chk2、cdc25a、cyclina、cycline、cdk2、p53和p21蛋白的表达情况。与对照组相比,随着配合物的加入,p53和p21蛋白的表达水平上升;而chk1、chk2、cdc25a、cyclina、cycline和cdk2蛋白的表达水平则受到抑制。实验结果表明配合物4和6可以通过抑制cyclin e/a
‑
cdk2复合物的活性来使bel
‑
7402细胞阻滞于g1期,从而抑制细胞增殖。该结果与流式细胞术检测的细胞周期实验一致。
[0252]
3.3.4流式细胞术检测配合物诱导肿瘤细胞凋亡
[0253]
annexin
‑
v
‑
fitc是一种荧光探针,可在钙离子存在的情况下与磷脂酰丝氨酸(ps)结合。当细胞发生早期凋亡时,ps开始由胞内转移到细胞膜外部,此时,annexin
‑
v可以与外翻的ps结合,可检测细胞的早期凋亡。细胞发生晚期凋亡时,pi(碘化吡啶)可以通过受损的细胞膜进而将dna染色,可检测细胞的晚期凋亡。因此,本节通过流式细胞术来检测配合物诱导细胞凋亡的百分数。
[0254]
图9是配合物4(0.4、0.8和1.6μm)作用bel
‑
7402细胞48h后,细胞凋亡的情况。加入配合物4后,细胞凋亡百分数分别是:9.76%、35.1%和55.8%,与对照组(3.41%)相比,凋亡百分数分别增加了:6.35%、31.69%和52.39%,凋亡百分数随加药量增加而增加,呈浓
度依赖关系。实验说明配合物4可以诱导bel
‑
7402细胞产生凋亡。
[0255]
图10是配合物6(0.35、0.7和1.4μm)作用bel
‑
7402细胞48h后,细胞凋亡的情况。加入配合物6后,细胞凋亡百分数分别是:27.1%、38.2%和60.6%,与对照组(0.595%)相比,凋亡百分数分别增加了:26.51%、37.61%和60.01%,凋亡百分数随加药量增加而增加。实验说明配合物6可以诱导bel
‑
7402细胞产生凋亡。
[0256]
3.3.5线粒体膜电位(δψm)的检测
[0257]
线粒体在细胞凋亡过程中发挥了关键作用(比如:影响细胞内atp水平,δψm的改变,线粒体膜通透性改变,活性氧的产生等)。线粒体外膜通透化是线粒体介导凋亡通路中的一个重要事件,它通过线粒体将细胞色素c(cyt
‑
c)等促凋亡蛋白释放到细胞质中,cyt
‑
c可以启动caspases依赖通路,从而裂解parp(poly adp
‑
ribose polymerase,一种dna修复酶,是caspases的切割底物),最终导致细胞凋亡。而线粒体膜电位(δψm)的破坏在线粒体膜通透性的增加中起着关键作用,被认为是线粒体介导凋亡的先决条件。
[0258]
图11和12分别是配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞作用48h后,细胞的细胞线粒体膜电位的变化情况。与对照组(1.88%)相比,bel
‑
7402细胞的δψ
m
都下降了:配合物4作用后,下降的百分比分别是:24.0%、36.7%、60.0%;配合物6作用后,下降的百分比分别是:34.7%、54.1%、65.9%;实验结果说明了配合物4和6都可以诱导bel
‑
7402细胞的δψ
m
下降,且有可能通过线粒体诱导bel
‑
7402细胞发生凋亡。
[0259]
3.3.6配合物对癌细胞内活性氧(ros)的影响
[0260]
细胞内活性氧(ros)水平升高作为氧化应激的信号,也可激活细胞凋亡信号通路。线粒体既是ros的靶细胞,也是产生更多ros的来源。一些研究表明,ros的产生会加剧线粒体膜电位的破坏,然后释放更多的细胞色素c(cyt c),从而导致caspase
‑
3裂解和最终的细胞凋亡。此外,活性氧可以与嘌呤碱、嘧啶碱和核糖相互作用,可以破坏dna的单链或双链,激活依赖dna的蛋白激酶和p53,导致细胞凋亡。
[0261]
图13和14分别是配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞作用48h后,细胞内ros的释放情况。通过荧光倒置显微镜观察发现:与对照组相比,配合物作用后,细胞发出的绿色荧光增强,说明配合物4和6均可引起bel
‑
7402细胞的ros水平升高,从而诱导细胞凋亡。
[0262]
3.3.7钙离子(ca
2+
)释放的检测
[0263]
线粒体介导的细胞凋亡的内在机制涉及线粒体及相关的线粒体蛋白,dna受损或者细胞中的癌基因过表达可以激活这一凋亡途径。同时,其他的一些因素(如:生长因子剥夺、ca
2+
水平上调、氧化剂和靶向药物等)也会使该通路被激活。我们已经知道,ca
2+
信号控制着许多重要的细胞功能,其在细胞内的稳态对于细胞的生存和死亡都极为重要。有研究表明,在细胞接收到凋亡信号时,钙离子由内质网向线粒体转移,使线粒体膜受损并增加膜通透性,从而导致细胞色素c(cyt c)和凋亡蛋白酶激活因子1(aapaf
‑
1)的释放,进而引起一系列导致细胞凋亡的关键事件,最终导致细胞凋亡。
[0264]
图15是配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞作用48h后,对细胞内ca
2+
的影响。流式细胞术实验结果表明:在配合物作用下,癌细胞内ca
2+
浓度增加,与对照组(黑色峰)相比,加入相应配合物后荧光增强(峰右移),ca
2+
释放水平提高,说明配合物4和6有可能是通过线粒体途径诱导bel
‑
7402细胞凋亡。
[0265]
3.3.8配合物激活肿瘤细胞中caspase
‑
3/9的表达
[0266]
凋亡细胞具有与病理坏死细胞不一样的形态特征,而细胞凋亡时大多数的形态学变化都是由一组半胱氨酸天冬氨酸特异性蛋白酶(caspases)引起的,这些蛋白酶在凋亡细胞中被特异性激活,由于caspases的活化是凋亡成功的关键,其家族成员在细胞凋亡的发生和执行中起着重要的作用。caspases家族分为两组:启动因子(caspase
‑
2,8,9和10)和执行因子(caspase
‑
3,6和7)。其中,caspase
‑
9是触发细胞凋亡的关键启动因子,细胞色素c(cyt
‑
c)从线粒体膜间间隙释放到细胞质后,结合凋亡蛋白酶激活因子
‑
1(apaf
‑
1)和procaspase
‑
9形成凋亡小体,促进caspase
‑
9的激活,活化的caspase
‑
9裂解并激活下游的caspases(如caspase
‑
3,caspase
‑
6,caspase
‑
7等),从而形成caspases级联反应来整合促凋亡信号,最终导致细胞凋亡。而caspase
‑
3则是细胞凋亡的关键执行者,被caspase
‑
9裂解并激活后,caspase
‑
3切割并裂解细胞内的数百种目标蛋白,最终导致细胞死亡。本节应用流式细胞术检测配合物5和7对癌细胞中caspase
‑
3/9表达的影响。
[0267]
应用流式细胞术检测配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞作用48h后,细胞中caspase
‑
3/9激活的情况。
[0268]
如图16所示,配合物4作用于细胞后,细胞中caspase
‑
3激活的百分数分别增加了15.22%、39.85%、47.16%。caspase
‑
9激活的百分数分别增加了18.32%、29.29%、62.69%。
[0269]
如图17所示,配合物6作用于细胞后,细胞中caspase
‑
3激活的百分数分别增加了10.43%、23.88%、38.57%。caspase
‑
9激活的百分数分别增加了13.35%、24.55%、36.16%。
[0270]
3.3.9western blot检测配合物对凋亡相关蛋白表达的影响
[0271]
线粒体功能障碍已被证明有助于细胞凋亡的发生,并且是凋亡通路的中心。该通路涉及三种关键的蛋白:b细胞淋巴癌
‑
2(bcl
‑
2)家族蛋白、caspases和线粒体促凋亡蛋白。bcl
‑
2家族蛋白通过调控线粒体膜的通透性,在线粒体介导的凋亡中起关键作用。bcl
‑
2家族蛋白中的bcl
‑
2蛋白是一种线粒体外膜蛋白,作为抗凋亡因子,bcl
‑
2蛋白过表达可以阻断细胞的凋亡过程。以bcl
‑
2等抗凋亡蛋白为靶点的分子治疗可启动癌细胞凋亡,比如我们知道的bcl
‑
2拮抗剂abt
‑
737可以有效地杀死白血病和淋巴瘤细胞。bax是一种促凋亡蛋白,在未激活时,bax蛋白可作为单体存在于胞浆内或与线粒体膜松散结合,但在活化过程中,bax向线粒体外膜移位并插入线粒体外膜。被激活的bax可以和bak形成同源低聚物,此聚合物参与线粒体膜孔的形成,并导致线粒体膜通透性增加,使细胞色素c(cyt c)释放到细胞溶胶中。cyt c与凋亡蛋白酶活化因子1(apaf
‑
1)及datp一起使启动因子procaspase
‑
9在凋亡小体内聚集并活化。随后,凋亡小体驱动caspases的活化,激活的caspases在整个细胞内裂解并使众多关键蛋白失效,最终导致细胞凋亡。
[0272]
如图18和19所示,配合物4(0.4、0.8和1.6μm)和6(0.35、0.7和1.4μm)对bel
‑
7402细胞作用48h后,能明显上调apaf
‑
1、bax、bak与cytochrome
‑
c蛋白的表达量,同时下调bcl
‑
2蛋白的表达量,实验结果表明:配合物4和6有可能是通过线粒体途径诱导细胞产生凋亡。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1