一种电压门控钠通道小分子抑制剂及其镇痛应用
1.本发明属于医药技术领域,具体涉及所述小分子化合物对电压门控钠通道(nav)的抑制活性及其在镇痛中的应用。
背景技术:
2.慢性疼痛严重困扰着全球近5亿人,截至今日还没有理想的治疗方法。阿片类药物是目前最为广泛使用的镇痛药物,但具有明显的副作用如成瘾和长期使用的耐药性等。电压门控钠通道(vgsc,nav)是一种选择性地转运钠离子的高度糖基化的整合膜蛋白。遗传学和药理学证据已证实电压门控钠通道(vgsc,nav)亚型nav1.7,nav1.8和nav1.9在背根神经节感受器神经元中表达,介导了疼痛信号向中枢的传递,因而是优秀的疼痛治疗靶点。靶向钠通道抑制剂的研发一直是国际疼痛领域研究的热点,在钠通道阻滞剂开发中最主要的挑战是亚型的选择性,这是因为nav亚型之间具有高序列同源性。理论上非选择性钠通道抑制剂可能导致一些不良副作用,特别是对心脏钠通道nav1.5和骨骼肌钠通道nav1.4的活性需尽可能避免。但选择性钠通道抑制剂的临床开发缓慢,一些国际制药大公司如genentech、 amgen和pfizer等每年都会投入大量资金和人力进行钠通道相关小分子抑制剂的筛选与研发。例如,genentech针对nav1.7通道开发的gx-936小分子;pfizer针对nav1.8通道开发的pf-01247324小分子。尽管针对钠通道结构域iv的pf/ca位点开发的nav1.7通道选择性小分子抑制剂在电生理水平具有强活性,但临床实验中表现不佳,可能是由于此类药物的神经渗透能力有限导致。目前临床上使用的靶向钠通道的药物包括局麻药,抗癫痫药,抗心率失常药和抗惊阙药物如利多卡因(lidocaine)、美西律(mexiletine)、卡马西平 (carbamazepine)以及阿米替林(amitriptyline)等都是非选择性钠通道抑制剂,作用于钠通道上结构保守的la位点。它们在对症剂量下的安全性已被多年的临床使用所证实,其中,阿米替林目前仍是治疗神经病理性疼痛的一线药物。因此,开发此类小分子钠通道抑制剂仍具有重大价值。
3.在本研究中,我们筛选到的有机小分子c65780是一种新的nav1.7
–
nav1.9通道拮抗剂。c65780由一个末端胺尾和四杂环头由一个短烃链连接组成。电生理实验分析表明, c65780将nav通道稳定在慢失活状态。在多种疼痛动物模型中c65780表现出于阿米替林相当的镇痛活性,且没有明显的急性毒性或运动抑制。因此,c65780是一个新的镇痛药物先导分子。
技术实现要素:
4.本发明提出了一种小分子化合物c65780对电压门控钠通道的抑制活性及其镇痛应用。
5.下面结合附图对本研究做进一步的说明。
附图说明:
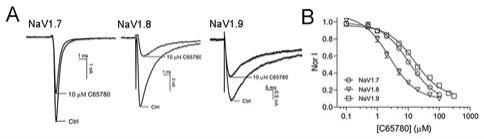
图1:c65780和amitriptyline的化学结构式;图2:c65780对异源表达的nav1.7、nav1.8、nav1.9通道电流的抑制活性:其中图a是c65780作用前后nav1.7、nav1.8、nav1.9通道的代表性电流,图b是在-100 mv钳制电压下c65780抑制nav1.7、nav1.8、nav1.9通道电流的浓效关系曲线;图3:c65780和阿米替林对异源表达nav1.7通道动力学的影响:其中图a是c65780作用前后nav1.7通道代表性电流;图b是c65780和阿米替林作用前后 nav1.7通道的i-v曲线;图c是c65780和阿米替林对nav1.7通道稳态激活的影响:图d 是c65780对nav1.7通道稳态快失活的影响;图e是c65780和阿米替林对nav1.7通道稳态慢失活的影响;图f是在-120mv和-60mv钳制电压下c65780和阿米替林抑制nav1.7通道的浓效关系;图4:c65780抑制大鼠背根部神经节(drg)神经元细胞上的navs通道:其中图a是在-120mv和-60mv钳制电压下c65780对drg神经元上navs电流的抑制作用;图b是在-120mv和-60mv钳制电压下c65780抑制drg神经元上navs电流的浓效关系曲线;图5:c65780在急性、炎症性和神经病理性疼痛模型中的镇痛活性:其中图a是c65780在小鼠醋酸扭体模型中的镇痛活性;图b是c65780在小鼠热板痛模型中的镇痛活性;图c是c65780在小鼠热板痛模型中给药60min后的镇痛活性分析;图d是 c65780在福尔马林致炎疼痛模型的镇痛活性;图e是c65780在福尔马林致炎疼痛模型中i 相和ii相痛的镇痛活性;图f,左:完全弗氏佐剂(cfa)疼痛模型造模前后小鼠回爪阈值 (pwt);右:c65780在小鼠cfa疼痛模型的镇痛活性;图g是c65780在cfa疼痛模型中给药60min后的镇痛活性分析;图h,左:坐骨神经部分结扎(sni)疼痛模型造模前后小鼠回爪阈值(pwt);右:c65780在sni疼痛模型中的镇痛活性;图i是c65780在sni 疼痛模型中给药60min后的镇痛活性分析。
7.图6:c65780的nav通道亚型选择性分析及其毒副作用评价:其中图a是c65780作用前后nav1.2、nav1.3、nav1.4和nav1.5通道的代表性电流;图b 是c65780抑制nav1.2、nav1.3、nav1.4和nav1.5通道电流的浓效关系曲线;图c是c65780和阿米替林抑制心肌钠通道nav1.5电流的浓效关系曲线;图d是c65780在小鼠强迫游泳实验中对小鼠运动能力影响的测定。
具体实施方式:
小分子化合物2-(2-(二乙基氨基)乙基)茚并[1,2,3-de]酞嗪-3(2h)-酮(简称c65780)由上海纳孚生物科技有限公司进行化学合成得到,技术路线是以3-芴酮-1-甲酸为母核与水合肼反应后形成中间产物茚并[1,2,3-de]酞嗪-3(2h)-酮,中间产物与二乙氨基乙基氯化物生成所述小分子化合物。其合成路径为为了揭示该小分子化合物的镇痛应用,利用膜片钳技术测定了c65780对nav1.7、
nav1.8、 nav1.9通道的抑制活性并探讨了其作用机制,利用动物疼痛模型验证了其镇痛活性,并对其毒副作用进行评估分析。
[0009]
1.主要材料和仪器全细胞膜片钳记录使用的记录采集数据放大器为epc10 usb amplifier(heka,elektronik, lambrecht,germany),数据记录和控制软件为patchmaster(heka,elektronik,lambrecht, germany)。其它实验仪器包括pc-10电极拉制仪(narishige,tokyo,japan)、日本 olympus ix70倒置显微镜、超净工作台、恒温细胞培养箱。实验用小鼠、大鼠均来自湖南斯莱克景达实验动物有限公司。
[0010]
2.实验方法和结果分析细胞转染:(1)培养hek-293t细胞,当细胞密度达到80-90%时,弃培养液,用1
×
pbs漂洗一次,加入 2ml无血清培养基(opti-mem)。(2)配制a液:250μl无血清培养基+4ug dna,轻轻混匀,室温静置5分钟。(3)配制b液:250μl无血清培养基+8μl lipfectamine 2000,轻轻混匀,室温静置5分钟。(4)将a液与b液轻轻混匀,室温下静置20min。(5)将dna-脂质体混匀后,轻轻滴加入细胞中。(6)4-6小时后,更换含有血清的全培养基。
[0011]
活性检测:实验前将细胞外液从冰箱取出恢复室温,更换培养皿内的培养液。更换溶液时动作要轻柔,防止细胞从培养皿底部脱落。倒置显微镜下选择细胞膜较为光滑、细胞质均匀的细胞,在室温20~25℃条件下进行膜片钳实验。选用硼硅酸盐玻璃毛细管为玻璃电极材料,玻璃电极在拉制仪(pc-10,narishige)上经两步拉制而成,拉制完成后在玻璃电极内灌细胞内液。玻璃电极入水电阻为1.5~2.5mω。待电极与细胞膜之间形成高阻抗的京欧(gω)封接后,补电极快电容,转换为全细胞记录模式,给予细胞一短促而有力的负压,将钳制在电极中的细胞膜迅速打破,再补偿细胞慢电容。将细胞钳制在相应测试电压下,细胞稳定4~6min开始记录电流。系统电阻(rs)在实验过程中始终保持在5~10mω的范围之内,基本维持不变,系统串联电阻(rseries compensation)补偿80%。
[0012]
2.1 c65780对异源表达的nav1.7、nav1.8、nav1.9通道的抑制活性nav1.7-1.9通道是决定背根神经节(drg)神经元动作电位去极化阶段的关键膜蛋白。因此,以nav1.8为靶通道筛选,得到小分子2-(2-(二乙基氨基)乙基)茚并[1,2,3-de]酞嗪-3(2h)-酮 (命名为c65780)可抑制其电流。c65780在结构上由一个胺尾和一个四杂环头由短烃链连接组成,该结构类似于三环抗抑郁药阿米替林,两者的胺尾也存在于局麻药的结构中(见图 1)。c65780也抑制nav1.7和nav1.9通道的电流(见图2a)。经过计算,在钳制电压为
‑ꢀ
100mv下,c65780抑制nav1.7、nav1.8、nav1.9通道电流的半数抑制浓度(ic
50
)分别为11.3
±
0.4μm、2.7
±
0.3μm和19.2
±
2.3μm(图2b,n=6-8)。
[0013]
2.2 c65780和amitriptyline对异源表达的nav1.7通道的动力学分析为了深入了解c65780与nav通道的作用机制,首先分析其对nav1.7通道的电流-电压关系 (i-v曲线)的影响。同时也将神经病理性痛的临床一线用药阿米替林纳入测试。如图3a所示,c65780抑制从-100mv到+100mv的一组去极化电压刺激下诱发的nav1.7通道的电流。但c65780和阿米替林并没有改变通道的峰值电压、翻转电位以及i-v曲线形状(见图 3b)。此外又对c65780和阿米替林作用前后nav1.7通道的电导-电压曲线(g-v曲线)进行分析(见图3c),其中对照组
的半数稳态激活电压(va)和通道激活斜率因子(ka)为-21.9
ꢀ±
1.4mv和5.5
±
0.6mv,经20μm c65780和20μm阿米替林处理后的va和ka分别为
‑ꢀ
21.1
±
2.2mv和5.6
±
0.4mv、-21.1
±
1.2mv和5.1
±
0.5mv(n=6-7),结果显示c65780和阿米替林并没有改变通道的va和ka。如图3d所示在c65780和阿米替林对nav1.7通道稳态快失活的影响中,对照组的半数稳态失活电压(vh)和通道失活斜率因子(kh)为-67.8
ꢀ±
3.3mv和-6.3
±
0.3mv,经20μm c65780或20μm阿米替林处理后的vh和kh分别为
‑ꢀ
84.6
±
2.1mv和-6.2
±
0.2mv、-84.2
±
3.4mv和-6.1
±
0.5mv(n=6-7),两者并未改变通道的 kh但分别使nav1.7通道快失活的半数稳态失活电压(vh)向超极化方向漂移约16.8mv和 16.4mv。如图3e所示,在对通道稳态慢失活的影响中,对照组的vh、kh和失活通道平台比例(p
non-inac
)为-57.6
±
3.9mv、-16.5
±
2.2mv和42.5
±
3.9%,经20μm c65780或20μm 阿米替林处理后的vh、kh和p
non-inac
分别为vh=-83.3
±
2.5mv和-89.5
±
2.8mv,kh=-6.1
±ꢀ
0.6mv和-7.7
±
1.2mv,p
non-inac
=23.2
±
3.6%和8.0
±
1.1%,两者显著将nav1.7通道稳态慢失活的vh向超极化方向漂移、改变kh值并降低失活通道平台比例。如图3f所示,其在-120 mv和-60mv钳制下c65780抑制nav1.7通道电流的ic
50
值为10.3
±
0.4μm和0.4
±
0.4μm,而阿米替林的对应值为29.8
±
1.3μm和1.0
±
0.5μm,两者在去极化钳制电压下对 nav1.7通道电流的抑制能力比在超极化电压下大大增加,提示药物均以更高的亲和力与失活状态通道结合。
[0014]
上述数据表明c65780和阿米替林都通过促进/稳定nav1.7通道的慢失活状态抑制其电流。
[0015]
2.3 c65780对大鼠背根部神经节(drg)神经元细胞上navs通道的抑制活性大鼠drg神经元富含多种navs通道,通过检测c65780对大鼠drg神经元navs通道的影响进一步探究c65780对navs通道的抑制活性。图4a为在超极化电压-120mv下和半失活电压-60mv下,c65780作用前后的drg神经元navs通道代表性电流。如图4b所示,在
‑ꢀ
120mv和-60mv钳制下c65780的ic
50
值分别为10.2
±
0.5μm和2.7
±
0.8μm,与其对 nav1.7通道的效果类似,在去极化钳制电压下药物对drg神经元navs通道电流的抑制能力较超极化钳制电压增强。
[0016]
2.4 c65780在动物急性或慢性疼痛模型中的镇痛活性该部分实验验证了c65780在三种急性疼痛模型和两种慢性疼痛模型的镇痛活性。其中急性疼痛模型包括小鼠醋酸扭体疼痛模型,热板疼痛模型和福尔马林炎症疼痛模型,慢性疼痛模型包括完全弗氏佐剂(cfa)疼痛模型和坐骨神经部分结扎(sni)模型。均分为三组,生理盐水对照组,阳性对照药物阿米替林组和测试药物c65780组。
[0017]
2.4.1醋酸扭体模型icr小鼠(18~22g)随机分组(n=6)。c65780溶解于无菌生理盐水采用灌胃方式给药,在灌胃 60min后,采用腹腔注射方式给小鼠注射200μl 0.8%的醋酸,计数小鼠注射乙酸后30min 内的扭体次数。阳性对照药物为15mg/kg阿米替林,对照组给予同样体积的无菌生理盐水 (saline)。实验数据,采用t检验比较各组的显著性差异。将各组的扭体次数标准化于对照组,计算出疼痛评分1、5、10、15mg/kg c65780组和15mg/kg阿米替林阳性对照组分别为 100.1%,83.1%,54.4%,38.3%和43%(n=6)。实验结果显示:c65780减轻疼痛反应的效果具有剂量依赖性,10mg/kg和15mg/kg c65780组的小鼠扭体次数明显少于生理盐水对照组。此外,15mg/kg c65780组的小鼠扭体次数与15mg/kg阿米替林阳性对照组不具有显著性差异,表明两者镇痛效果相当(n=6)。
[0018]
2.4.2热板疼痛模型将c57bl/6小鼠做实验前筛选,剔除对热不敏感和对热过于敏感的个体,选择能够受热 5-30s的个体进行实验,并求出基础疼痛阈值。放置小鼠于55
±
1℃的恒温板上,观察记录小鼠从放入到舔后足所需时间,以此作为疼痛阈值。小鼠随机分为六组(n=6)。c65780溶解于无菌生理盐水配制成1、5、10、15mg/kg c65780组,采用灌胃方式给药,分别在注射后 30min,60min,90min和120min测定小鼠的痛阈。15mg/kg阿米替林作为阳性对照组,空白对照组给予同样体积的无菌生理盐水(saline)。实验数据,采用t检验比较各组的显著性差异。。在药物镇痛时程图中,阿米替林组和c65780组在60min时均基本达到最大镇痛效果,其镇痛活性在60min后开始下降。图5c为60min时的镇痛活性的柱状图,生理盐水对照组、1、5、 10、15mg/kg c65780组和15mg/kg阿米替林组分别为11.4
±
0.6s,11.9
±
0.9s,13.9
±
0.9s,16.2
ꢀ±
0.8s,17.8
±
0.9s和16.9
±
1.0s。该实验结果显示c65780的止痛效果也是具有剂量依赖性的,并在给药后1小时达到峰值效果,与生理盐水对照组相比,10mg/kg和15mg/kg c65780 以及15mg/kg阿米替林阳性对照组在60min时对热刺激的反应显著降低,且15mg/kgc65780的镇痛效果略优于15mg/kg阿米替林阳性对照组。
[0019]
2.4.3福尔马林炎症疼痛模型经随机分组的icr小鼠在给药60min后足底注射5%福尔马林溶液,该疼痛模型分为两个时相,包括i相伤害性感受疼痛期和ii相炎性反应疼痛期。图5d为福尔马林致炎模型中,随着时间变化的小鼠平均舔足时间曲线图。图5e左图为i相疼痛期,生理盐水组小鼠的平均舔足时间为147.2
±
8.9s,1、5、10、15mg/kg c65780组和15mg/kg阿米替林阳性对照组平均舔足时间分别为133.3
±
9.1s,118.2
±
8.2s,82.8
±
6.9s,69.7
±
6.5s和81.2
±
9.2s;右图为 ii相疼痛期,生理盐水组小鼠的平均舔足时间为250.3
±
14.3s,1、5、10、15mg/kg c65780 组和15mg/kg阿米替林阳性对照组平均舔足时间分别为235.2
±
13.9s,158.2
±
13.5s,120.3
±ꢀ
11.0s,96.2
±
10.6s和100.5
±
12.2s。实验结果显示:c65780呈现剂量依赖性的减少i期和ii 期的疼痛反应;在i期中,10mg/kg和15mg/kg c65780以及15mg/kg阿米替林阳性对照组显著降低了小鼠的舔足时长;在ii期中5、10、15mg/kg c65780以及15mg/kg阿米替林阳性对照组显著降低了小鼠的舔足时长。且15mg/kg c65780的镇痛效果与15mg/kg阿米替林阳性对照组相当(n=6)。
[0020]
2.4.4完全弗氏佐剂(cfa)疼痛模型经筛选后的c57bl/6小鼠右后足底注射30μl cfa诱导慢性疼痛,24小时后使用von frey细纤维丝评估机械敏感性并随机分组。图5f右图所示,在造模前后小鼠的平均足底疼痛阈值 (pwts)为1.18g和0.04g;左图显示c65780以剂量和时间依赖的方式减少cfa诱导的痛觉,在给药后60min达到最高值。图5g的柱状图显示在给药60min时的小鼠pwts,生理盐水对照组、1、5、10、15mg/kg c65780组和15mg/kg阿米替林组分别为0.04g,0.08g,0.18 g,0.28g,0.5g和0.47g,5、10、15mg/kg c65780以及15mg/kg阿米替林阳性对照组显著高于生理盐水对照组,且15mg/kg c65780的镇痛效果与15mg/kg阿米替林阳性对照组相当 (n=6)。
[0021]
2.4.5坐骨神经部分结扎(sni)模型图5h右图所示,在造模前小鼠的平均足底疼痛阈值(pwts)为1.21g;在手术后14天时平均足底疼痛阈值(pwts)为0.37g。图5h左图显示,c65780剂量依赖性地降低造模后小鼠
的疼痛反应,其作用持续约两小时。图5i的柱状图显示在给药60min时的小鼠pwts,生理盐水对照组、1、5、10、15mg/kg c65780组和15mg/kg阿米替林组分别为0.36g,0.47g,0.63 g,0.77g,0.94g和0.89g,10、15mg/kg c65780以及15mg/kg阿米替林阳性对照组镇痛效果显著高于生理盐水对照组,且15mg/kg c65780的镇痛效果与15mg/kg阿米替林阳性对照组相当(n=6)。
[0022]
在醋酸、热和福尔马林致炎剂等引起的急性疼痛模型中以及cfa完全弗氏佐剂诱导的慢性炎性疼痛模型和sni神经结扎导致的慢性神经痛模型中,c65780表现出了良好的镇痛效果且具有浓度依赖性,并与相对应浓度下的阿米替林相当。
[0023]
2.5c65780副作用的评价如图6a所示,c65780可以剂量依赖性地抑制nav1.2-1.5电流。在钳制电压为-100mv下, c65780对nav1.2、nav1.3、nav1.4和nav1.5通道的半数抑制浓度(ic
50
)分别为13.9
±ꢀ
2.7μm,6.0
±
0.3μm,24.5
±
2.8μm和4.1
±
0.5μm(见图6b,n=6-8)。如图6c所示,比较c65780和阿米替林对心脏钠通道nav1.5的抑制作用,在钳制电位为-120mv下,c65780 和阿米替林的ic
50
分别为16.7
±
1.8μm和6.4
±
0.5μm。与阿米替林相比,c65780对nav1.5 通道的活性稍弱。尽管对钠通道具有广泛的抑制作用,c65780在急性毒性实验中用15 mg/kg c65780灌胃小鼠持续观察记录15天,实验小鼠并没有表现出明显的毒性作用。此外,在1、5、10和15mg/kg c65780给药后小鼠进行强迫游泳试验,也并未观察到药物对实验小鼠运动功能的抑制(见图6d,n=6)。因此,推断c65780与阿米替林具有相似安全性。
[0024]
综上,小分子化合物c65780将nav通道稳定于慢失活状态,其作用机制与临床使用的神经病理性痛药物阿米替林类似,且镇痛活性与阿米替林相当。其对心脏钠通道的抑制活性较阿米替林稍弱。同时,小鼠急性毒性实验和强迫游泳实验中c65780无明显的急性毒性和运动抑制的副作用。因此,c65780是一个新的镇痛药物先导分子,具有开发成为新型镇痛药物的潜力。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1