靶向2019新型冠状病毒的木瓜蛋白酶样蛋白酶PLpro的稳定多肽类蛋白共价抑制剂
靶向2019新型冠状病毒的木瓜蛋白酶样蛋白酶plpro的稳定多肽类蛋白共价抑制剂
技术领域
1.本发明属于生物工程领域,涉及一种稳定多肽类蛋白共价抑制剂,具体来说是一种靶向2019新型冠状病毒的木瓜蛋白酶样蛋白酶plpro的稳定多肽类蛋白共价抑制剂及用途。
背景技术:
2.covid
‑
19病原体是一种新型冠状病毒,称为sars
‑
cov
‑
2。这种冠状病毒具有高传染性和致病性,自2019年12月首次发现以来在世界上广泛传播,迄今为止已经导致超过2亿人感染,死亡人数超过400万。covid
‑
19感染的潜伏期为2
‑
14天,最长可达24天。这些较长的潜伏期,因其可传播性和无症状性质,是造成大量感染的原因。越来越多的covid
‑
19病例反映了当前形势的严重性,需要有效的解决方案,但目前仍缺乏有效的抗病毒治疗。
3.目前研究表明这种病毒与sars
‑
cov相似。一个有潜力的抗病毒药物靶点是sars
‑
cov
‑
2编码的半胱氨酸蛋白酶
‑‑
木瓜蛋白酶样蛋白酶plpro。sars
‑
cov
‑
2的复制酶基因编码两个蛋白pp1a和pp1ab,之后将这两种多蛋白加工成16种非结构蛋白(nsps),以实现基因复制和rna的转录。蛋白水解通过两种半胱氨酸蛋白酶
‑‑
木瓜蛋白酶样蛋白酶(plpro)和主要蛋白酶(mpro/3clpro)。plpro通过识别保守序列lxgg从多蛋白n末端释放nsp1
‑
nsp3,还可以催化去除宿主细胞蛋白上的k48连接的泛素,并从宿主蛋白中去除干扰素刺激基因15(isg15),从而干扰宿主的免疫应答。
4.长期以来人们关注的治疗性药物主要集中在两类:小分子药物(small molecules)、蛋白类药物(biologics)。小分子药物靶向的化学空间具有一定的局限性,蛋白类药物存在稳定性差以及无法穿透细胞膜,这两类治疗性药物由于其自身生物物理性质的局限性,并不能有效覆盖这所有已确证的重要分子靶点。
5.多肽类药物则是另一类引起人们广泛关注和兴趣的靶向分子。与生物大分子类似,多肽类分子对于靶点也有较高的结合力与选择性,相对于小分子类药物具有更小的脱靶效应。而多肽在体内的代谢产物为氨基酸,最大限度地降低了毒性。传统的多肽类药物由于氨基酸残基数目有限,无法有效形成复杂二级结构,在生理溶液中有很高自由度并呈无规则线性状态,不仅降低了其特异性同时容易被蛋白酶所降解。并且多肽类药物的细胞膜穿透能力不是很好。通过化学手段修饰多肽将其稳定成为有二级结构的构象,不仅能够增加其对蛋白酶的稳定性,还能增强多肽的细胞膜穿透能力,并且还能够通过降低多肽与靶点结合时的熵变从而提高多肽与靶点的结合能力。通过各种化学修饰手段,将参与多种蛋白
‑
蛋白相互作用的二级结构单元提取出来进行修饰,不仅通过稳定他们的二级构象来模拟原始蛋白质的相互作用,更重要的是通过修饰可以使得这些蛋白质二级结构单元具备穿透细胞膜的能力,进而靶向细胞内蛋白
‑
蛋白相互作用。我们课题组在2019年最近通过cys和met之间的双烷基化开发了一种肽环化策略,以多肽侧链上的硫盐作为新的弹头在多肽配体的诱导下与蛋白cys在空间靠近的条件下发生共价反应。基于此,本课题设计了一系列
靶向plpro的锍盐稳定肽,这些肽可以与plpro共价结合并抑制plpro的活性,并且在细胞内从宿主蛋白中去除干扰素刺激基因15(isg15),从而干扰宿主的免疫应答。
技术实现要素:
6.针对现有技术中的上述技术问题,本发明提供了一种靶向2019新型冠状病毒蛋白plpro的稳定多肽类蛋白共价抑制剂及用途,所述的这种靶向2019新型冠状病毒蛋白plpro的稳定多肽类蛋白共价抑制剂及用途要解决现有技术中的药物对于治疗新型冠状病毒引起的肺炎的效果不佳的技术问题。
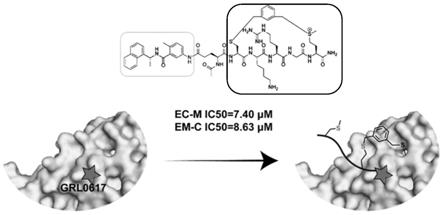
7.本发明提供了一系列靶向2019新型冠状病毒蛋白plpro的稳定多肽类蛋白共价抑制剂,其结构式如下所示,
8.或者或者或者或者或者或者或者或者或者或者
9.进一步的,其氨基酸序列分别是:
10.多肽序列连接小分子 ecmgrl0617
‑
e
‑
cyclic(clrgm)1,3
‑
二溴甲基苯seq id no.1所示emcgrl0617
‑
e
‑
cyclic(mlrgc)1,3
‑
二溴甲基苯seq id no.2所示elrgggrl0617
‑
elrgg1,3
‑
二溴甲基苯seq id no.3所示cm1ac
‑
lrgg
‑
cyclic(caaam),1,3
‑
二溴甲基苯seq id no.4所示cm2ac
‑
lrgg
‑
cyclic(maaac)1,3
‑
二溴甲基苯seq id no.5所示cm3ac
‑
cyclic(mrggc)1,3
‑
二溴甲基苯seq id no.6所示cm4ac
‑
cyclic(crggm)1,3
‑
二溴甲基苯seq id no.7所示cm5ac
‑
l
‑
cyclic(mggc)1,3
‑
二溴甲基苯seq id no.8所示
cm6ac
‑
l
‑
cyclic(cggm)1,3
‑
二溴甲基苯seq id no.9所示cm7ac
‑
cyclic(mlrgc)1,3
‑
二溴甲基苯seq id no.10所示cm8ac
‑
cyclic(clrgm)1,3
‑
二溴甲基苯seq id no.11所示
11.本发明还提供了上述的稳定多肽类蛋白共价抑制剂在制备用于抑制蛋白plpro酶活的药物中的用途。
12.本发明还提供了上述的稳定多肽类蛋白共价抑制剂在制备用于靶向2019新型冠状病毒蛋白plpro的药物中的用途。
13.本发明还提供了上述的稳定多肽类蛋白共价抑制剂在制备用于治疗新型冠状病毒引起的肺炎的药物中的用途。
14.本发明提供了一种靶向2019新型冠状病毒(sars
‑
cov
‑
2)的木瓜蛋白酶样蛋白酶(papain
‑
like protease,plpro)的稳定多肽类蛋白共价抑制剂,其氨基酸序列衍生于plpro识别的保守序列lrgg。酶活实验充分证明了该共价多肽抑制剂有效降低了木瓜蛋白酶样蛋白酶plpro的活性。
15.本发明采用了多肽上甲硫氨酸
‑
半胱氨酸与双烷基化试剂反应形成单个锍盐的方法来稳定靶向plpro的锍盐环肽。采用之前文献报道的基于锍盐稳定的多肽配体对蛋白半胱氨酸进行选择性共价修饰方法,当该环肽配体与靶点蛋白plpro相互识别时,环肽上的锍盐与蛋白plpro半胱氨酸在空间靠近下发生亲核反应实现对蛋白的共价修饰。通过sds
‑
page分析和质谱分析证明该多肽通过与靶蛋白相互识别后与靶蛋白plpro的c111发生共价反应。
16.本发明通过荧光偏振检测、酶活检测等实验,证实该多肽可以很好的结合plpro蛋白,并通过多肽上的锍盐与蛋白plpro上的半胱氨酸发生共价反应,阻断plpro蛋白与底物分子lrgg
‑
amc的结合。本发明还通过流式细胞分析、细胞存活等实验,证实该多肽可以很好的进入hct116、a549等细胞并不会引发细胞大规模死亡。
附图说明
17.图1为锍盐共价抑制剂多肽分子合成方式图(以cm2和emc为例)。
18.图2为多肽cm1
‑
8与plpro蛋白共价结合图谱。
19.图3为多肽emc与plpro蛋白和plpro
c111s
共价结合图谱。
20.图4为多肽ecm与plpro蛋白共价结合一级质谱图。
21.图5为多肽cm1
‑
8对plpro酶活抑制效率图。
22.图6为多肽emc、emc、elrgg和grl0617对三种plpro酶活抑制效率图。
23.图7为isg15免疫印迹验证多肽恢复细胞isg化水平。
24.图8为不同多肽对人非小细胞肺癌细胞a549和正常细胞hek293t增殖抑制能力。
25.图9是锍盐稳定多肽共价抑制plpro示意图。
具体实施方式
26.下面结合附图对本发明进一步说明。
27.实施例1
28.本发明采用之前文献报道(d.wang,m.yu,et al.chem.sci.10,4966
‑
4972)的锍盐
稳定多肽技术,通过多肽上甲硫氨酸与半胱氨酸与双烷基化试剂反应形成锍盐环肽,既能稳定多肽,又能与靶蛋白相互作用位点上的半胱氨酸发生共价修饰,阻断木瓜蛋白酶样蛋白酶plpro和病毒多种非结构蛋白的结合,抑制病毒基因的复制。
29.本发明提供了一系列靶向2019新型冠状病毒(sars
‑
cov
‑
2)的木瓜蛋白酶样蛋白酶(papain
‑
like protease,plpro)的稳定多肽类蛋白共价抑制剂。发明人合成了多条不同的多肽,如表一所示。
30.表一:不同靶向新冠病毒蛋白plpro的稳定多肽类蛋白共价抑制剂分子序列。
31.多肽序列连接小分子分子量ecmgrl0617
‑
e
‑
cyclic(clrgm)1,3
‑
二溴甲基苯1137.54emcgrl0617
‑
e
‑
cyclic(mlrgc)1,3
‑
二溴甲基苯1137.54elrgggrl0617
‑
elrgg1,3
‑
二溴甲基苯858.50cm1ac
‑
lrgg
‑
cyclic(caaam),1,3
‑
二溴甲基苯1421.60cm2ac
‑
lrgg
‑
cyclic(maaac)1,3
‑
二溴甲基苯1421.60cm3ac
‑
cyclic(mrggc)1,3
‑
二溴甲基苯1095.41cm4ac
‑
cyclic(crggm)1,3
‑
二溴甲基苯1095.41cm5ac
‑
l
‑
cyclic(mggc)1,3
‑
二溴甲基苯1052.60cm6ac
‑
l
‑
cyclic(cggm)1,3
‑
二溴甲基苯1052.60cm7ac
‑
cyclic(mlrgc)1,3
‑
二溴甲基苯1151.47cm8ac
‑
cyclic(clrgm)1,3
‑
二溴甲基苯1151.47
32.实施例2多肽的制备及分离纯化步骤:
33.本发明的多肽按照氨基酸序列固相合成,为常规技术,在此不再赘述,本实施例仅描述制备上述稳定多肽的核心步骤如下(以cm1为例):
34.具体操作步骤(图1)为:
35.(1)多肽固相合成:称取rink amide mbha树脂于接肽管中,加入二氯甲烷(dcm),鼓氮气溶胀30min。加入50%(v/v)吗啡啉的n,n
‑
二甲基甲酰胺(dmf)溶液,鼓氮气30min,脱去fmoc保护基团。用dmf和dcm交替洗涤树脂之后,将配好的fmoc
‑
aa
‑
oh(5eq,0.4m,dmf)溶液,6
‑
氯苯并三氮唑
‑
1,1,3,3
‑
四甲基脲六氟磷酸酯(hctu)(5eq,0.38m,dmf)溶液,n,n
‑
二异丙基乙胺(dipea)(10eq)混匀后加入树脂中鼓氮气1h。抽掉反应液,按上述方法洗树脂后进行下一步操作。接下来的氨基酸与上述方法相同。多肽的n端用乙酸酐和dipea乙酰化(1:1.8摩尔比),溶于dcm)30分钟(两遍)。
36.(2)分子内关环:在树脂上将半胱氨酸(cys)侧链保护基的trt基团脱保护(脱保护液:tfa/tis/dcm=3/5/92,摩尔比),分次脱去直至溶液不再变黄,之后分别用dmf、dcm交叉洗涤各五次,1,3
‑
二(溴甲基)苯试剂(2当量)和dipea(4当量)溶于dmf后加入树脂中反应2小时。
37.(3)多肽纯化:用三氟乙酸(tfa),三异丙基硅烷(tips)和h2o(v:v:v=9.5:0.25:0.25)剪切液将多肽从树脂上切下来,除去剪切液。用高效液相色谱进行纯化分离,最后质谱(ms)确认分子量。得到纯的多肽分子,具体结构式如前述。
38.实施例3多肽分子与蛋白plpro体外共价
39.不同带有荧光基团的多肽上甲硫氨酸与半胱氨酸与双烷基化试剂反应形成锍盐
环肽分别与蛋白共孵育,蛋白与锍盐多肽发生共价反应,可看到蛋白条带上有荧光显示(图2),说明cm1
‑
8多肽可以与plpro蛋白发生共价反应,而一般的线性肽则无法产生共价连接。
40.我们选择肽em
‑
c作为例子来研究反应动力学和化学计量研究。fam标记肽em
‑
c(10μm)与sars
‑
cov
‑
2plpro(5μm)在pbs缓冲液中反应不同时间(0.5、1、2、3、4小时)。该反应表现出剂量依赖性。动力学和化学计量研究清楚地显示了结合的高效率(图3a、b)。
41.多肽具有较好的特异性,主要与plpro的c111位的半胱氨酸进行共价反应,将plpro的111位半胱氨酸突变为丝氨酸,多肽基本不与plpro发生共价反应(图3c)。
42.为了评估肽em
‑
c和ec
‑
m在复杂蛋白质组环境中标记plpro的能力,在293t细胞裂解物(300μg)中加入plpro(5μm),然后用fam标记肽em
‑
c和ec
‑
m(10μm)处理。凝胶数据显示具有正确分子量的清晰单一荧光条带,表明肽em
‑
c和ec
‑
m与plpro干净且选择性地缀合(图3d)。
43.此外,我们通过二级质谱验证了肽和蛋白质的结合位点。从sds凝胶上切下的胰蛋白酶消化蛋白质样品的ms/ms分析清楚地显示了来自ecm和plpro缀合的肽片段在cys111。这些结果表明肽ecm主要与plpro的c111共价标记(图4)。
44.实施例4多肽分子抑制plpro酶活实验
45.sars
‑
cov
‑
2plpro经测试可以切割底物lrgg
‑
acc使其荧光基团acc释放,使其荧光强度增加。使用不同浓度的n端乙酰化的多肽cm1
‑
8(0
‑
800μm),与plpro蛋白(0.1μm)在实验缓冲液(5mm nacl,20mm tris,5mm dtt,ph=8.0)中混合,在37℃水浴下反应1小时,之后加入底物lrgg
‑
acc(1μm),在黑底96孔板中使用酶标仪进行测量荧光发射强度(λ
ex
:355nm,λ
em
:460nm)。硫盐稳定肽不能有效抑制sars
‑
cov
‑
2plpro(图5)。
46.不含grl0617的硫盐稳定肽不能有效抑制sars
‑
cov
‑
2plpro,而硫盐稳定肽和grl0617缀合物ecm、emc具有更好的抑制能力。但是比grl0617的抑制效果略差(图6a)。通过不同浓度的多肽抑制剂对plpro的酶活抑制实验证明,抑制效果呈现剂量依赖性。此外,测试了肽
‑
药物偶联物对sars
‑
cov plpro和mers plpro的抑制能力。sars
‑
cov
‑
2plpro和sars
‑
cov plpro之间具有高序列同一性(83%),emc,ecm多肽同样可以抑制sars
‑
cov plpro,线性肽elrgg的抑制作用弱于硫盐稳定肽(图6b)。grl0617不能抑制mers plpro,与之前的文献结果一致,同样的ecm、emc和elrgg对mers plpro也没有抑制效果(图6c)。
47.实施例5多肽分子对细胞内plpro的去isg化的影响
48.采用细胞内基于isg15免疫印迹的方法来观察是否可以恢复被plpro抑制的细胞isg化水平,研究ecm和emc对细胞内plpro去isg化活性的抑制作用。与其在两种药物筛选试验中的活性一致,肽
‑
药物偶联物可以在基于细胞的方法中以剂量依赖性方式恢复细胞isg化水平,表明这两种肽
‑
药物偶联物都可以进入细胞抑制sars
‑
cov
‑
2plpro。同时,ecm比emc显示出更高的恢复细胞isg化水平的效力(图7)。
49.实施例6多肽对细胞存活的影响
50.为了评估该多肽ecm、emc和elrgg对不同细胞的杀伤能力,选择了癌细胞人非小细胞肺癌细胞a549和正常细胞hek293t来排除多肽库的非特异性毒性。
51.细胞活力通过mtt测定法。细胞在96孔板中以4
×
103接种,用溶于培养基(5%血清)的多肽处理24h,将mtt加入培养基孵育4h。然后加入dmso溶解沉淀物,采用酶标仪在490nm测定吸光度。其中未处理的细胞存活率为100%。
52.结果表明,ecm、emc和elrgg对癌细胞人非小细胞肺癌细胞a549和正常细胞hek293t基本没有毒副作用(图8)。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1