一种钆布醇中间体及制备方法和在制备钆布醇中的应用与流程
1.本发明涉及医药中间体合成技术领域,具体涉及一种钆布醇中间体及制备方法和在制备钆布醇中的应用。
背景技术:
2.钆布醇(gadobutrol)是一种细胞外分布、非组织特异的电中性非离子型水溶性含钆对比剂(gadolinium
‑
based contrast agent,简称gbca),它能增强组织的磁共振成像(magnetic resonance imaging,简称mri)造影效果,常用于脑、脊髓、肝脏等身体部位的mri检查。钆布醇具有弛豫性高、稳定性好、黏滞性及渗透性较低等优点,主要经肾小球滤过清除,毒性低,其高浓度制剂在mri成像中独具优势。
3.钆布醇的分子式为c18h31gdn4o 9,它是由钆(ⅲ)离子与大环配体2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸(butrol)构成的络合物,其分子结构如下:
[0004][0005]
拜耳开发出的1.0mol/l高浓度钆布醇水溶液,以gadovist为商品名,于1998年2月作为核磁共振造影剂首次在瑞士获批上市。在之后的数年里,gadovist又陆续在澳大利亚、加拿大、欧盟、美国等100多个国家和地区获批上市。gadovist在美国上市的适应症为成人和儿童(包括新生儿)血脑屏障(bbb)和或中枢神经系统造影、恶性乳腺疾病造影。钆布醇是欧洲指南推荐的3个低风险钆类造影剂之一,也是fda批准可用于新生儿的唯一一种钆类造影剂。
[0006]
由于钆布醇在影像诊断,特别是在mri诊断上的重要性,开发更为实用性的合成工艺也变得尤其迫切。
[0007]
最接近现有技术(inorg.chem.1997,36,6086
‑
6093)中详细阐述了制备钆布醇的方法,采用轮环滕宁的dmf
‑
dma三保护合成路线。以轮环滕宁为起始原料,经dmf
‑
dma三保护保护,再与4,4
‑
二甲基
‑
3,5,8
‑
三氧杂双环辛烷等比例反应,然后脱去甲酰保护基,随后与氯乙酸钠反应进行三取代,同时也脱去缩酮保护基,最后与三氧化二钆反应得到钆布醇。
[0008][0009]
但根据发明人研究发现,上述合成的路线存在五个比较严重的问题:第一,主料轮环藤宁价格昂贵,每公斤单价高于5500元;第二,第二步所采用的试剂4,4
‑
二甲基
‑
3,5,8
‑
三氧杂双环[5,1,0]辛烷价格极其昂贵,每公斤单价高于25000元;第三,反应原子经济性差,共经历了两次三步的保护/脱保护反应,同时也产生了大量三废;第四,反应收率较低,文献中制备百克级钆布醇的收率仅为46%,导致整条路线的经济性较差;第五,所制备得到的钆布醇纯度较低,达不到临床应用的99.9%以上的高纯度要求,该低纯度钆布醇还需要经后续一系列进一步的纯化措施才能将纯度提升到符合临床应用的要求,后续纯化成本高。这些问题都严重限制了该工艺的进一步应用,也使得该产品的公斤级和百公斤级放大变得比较困难。
[0010]
因此,以较低的物料成本和工艺成本,制备高纯度、高收率的钆布醇原料药仍是亟待解决的问题。
技术实现要素:
[0011]
鉴于上述现有技术中存在的问题,本发明提供了一种钆布醇中间体及制备方法和在制备钆布醇中的应用,用于解决现有技术中的钆布醇的合成工艺中存在的经济性差、成本较高,产品纯度低和收率低等问题。
[0012]
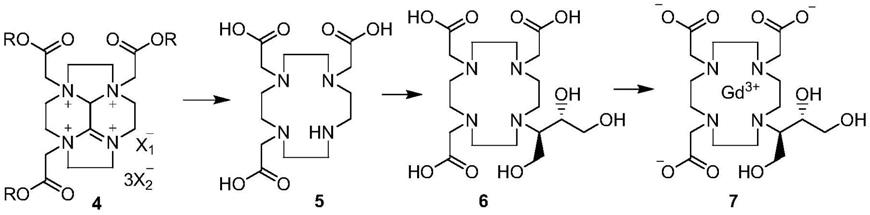
本发明的目的之一是提供一种由下述化学式4表示的钆布醇中间体:
[0013][0014]
式中,x1为氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基;x2为氯、溴或碘;r为钠、钾、甲基、乙基、异丙基、叔丁基或氢。
[0015]
本发明的目的之二是提供一种钆布醇中间体的制备方法,包括如下步骤:
[0016][0017]
(1)以式1化合物三乙烯四胺为原料,在惰性气体保护下,与第一关环试剂反应,反应温度为20~200℃反应,得到式2化合物,其中,所述的第一关环试剂是原甲酸三甲酯、原甲酸三乙酯、甲酸甲酯、甲酸乙酯、甲酸、dmf
‑
dma中的一种或者多种;
[0018]
(2)式2化合物与第二关环试剂反应,反应温度为20~200℃反应,得到式3化合物,其中,x1为氯、溴、碘、对甲苯磺酰氧基、甲磺酰氧基,所述的第二关环试剂是1,2
‑
二氯乙烷、1,2
‑
二溴乙烷、1,2
‑
二碘乙烷、1,2
‑
二对甲苯磺酰氧基乙烷,1,2
‑
二甲磺酰氧基乙烷,1
‑
氯
‑2‑
溴乙烷中的一种或者多种;
[0019]
(3)式3化合物与取代试剂发生取代反应,反应温度为20~200℃反应,得到式4化合物,式中,x1为氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基;x2为氯、溴或碘;r为钠、钾、甲基、乙基、异丙基、叔丁基或氢,所述取代试剂是氯乙酸、氯乙酸钠、氯乙酸钾、氯乙酸乙酯、氯乙酸叔丁酯、溴乙酸、溴乙酸钠、溴乙酸钾、溴乙酸甲酯、溴乙酸乙酯、溴乙酸叔丁酯、碘乙酸、碘乙酸钠、碘乙酸钾、碘乙酸乙酯或碘乙酸叔丁酯中的一种或者多种。
[0020]
具体的,在本发明一种钆布醇中间体的制备方法中,步骤(1)中所述第一关环试剂与式1化合物的物质的量比为1~10:1;步骤(2)中所述的第二关环试剂与式2化合物的物质的量比为:0.5~5:1;步骤(3)中所述取代试剂与式3化合物的物质的量比为1~10:1。
[0021]
本发明的目的之三是提供一种钆布醇的制备方法,包括上述钆布醇中间体的制备方法,还进一步包括如下步骤:
[0022][0023]
(4)以式4化合物为原料,在碱性试剂条件下水解,再经酸性试剂处理,得到式5化合物,式中,x1为氯、溴、碘、对甲苯磺酰氧基或甲磺酰氧基;x2为氯、溴或碘;r为钠、钾、甲基、乙基、异丙基、叔丁基或氢;
[0024]
(5)式5化合物与2,3
‑
环氧基
‑
1,4
‑
丁二醇进行开环加成反应,得到式6化合物;
[0025]
(6)式6化合物与钆试剂进行络合反应,再用树脂处理,得到式7化合物钆布醇。
[0026]
具体的,根据本发明的钆布醇的方法,进一步包括:
[0027]
步骤(4)中,所述的碱性试剂是氢氧化钠、氢氧化钾、氢氧化锂中的一种或者多种,所述的酸性试剂是盐酸、硫酸、磷酸、001
×
7树脂、amberlite ir
‑
120树脂、dowex 50
‑
x8树脂、lewatit s
‑
100树脂或diaion sk
‑
1b树脂中的一种或者多种,所述的碱性试剂与式4化合物的重量比为:0.1~10:1,所述的酸性试剂与式4化合物的重量比为:0.1~10:1;
[0028]
步骤(5)中,所述的2,3
‑
环氧基
‑
1,4
‑
丁二醇与式5化合物的物质的量比为:0.5~5:1;
[0029]
步骤(6)中,钆试剂是三氯化钆、三氧化二钆、乙酸钆中一种或者多种,所述的树脂是d301弱碱性阴离子树脂、d941弱碱性阴离子交换树脂、a100弱碱阴离子树脂中的一种或者多种,所述钆试剂与式6化合物的物质的量比为0.3
‑
5,所述树脂与式6化合物的重量比为0.1
‑
5:1。
[0030]
具体的,根据本发明上述化学式4表示的钆布醇中间体的制备钆布醇的方法,进一步包括使用滤膜过滤的过程和重结晶的过程。
[0031]
本发明的目的之四提供一种上述化学式4表示的钆布醇中间体在制备钆布醇中的应用,上述化学式4表示的钆布醇中间体可用做钆布醇的中间体或参照物,还可用于钆布醇的分析检测和杂质研究。
[0032]
与现有技术相比,本发明具有如下有益效果:本发明路线设计合理,原料价格较低,总收率高,产品纯度高,产品纯化成本低,经济性良好,能充分满足产品工业化生产的需求。
具体实施方式
[0033]
下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述地实施例仅仅是本发明的一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围.
[0034]
实施例1:
[0035][0036]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0037]
保持氮气微正压,向2l反应瓶中依次加入600ml 2
‑
甲基四氢呋喃,146.24g(1.0mol)式1化合物三乙烯四胺和222.85g(2.1mol)原甲酸三甲酯,加完后,搅匀;反应液升温至80℃反应,此时反应液出现回流。反应液保温80℃继续反应5小时,反应完成。
[0038]
反应液冷却析晶得158.25g(0.952mol)白色固体状产品。
[0039]
收率95.2%,hplc纯度99.2%,1h nmr(400mhz,cdcl3):δ6.72(s,2h),3.74(7,
4h),3.11
‑
3.24(m,8h)。
[0040]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓溴盐的制备
[0041]
向2l反应瓶中依次加入600ml乙腈,158.27g(0.952mol)式2化合物,183.44g(0.976mol)1,2
‑
二溴乙烷和243.89g(1.765mol)碳酸钾,加完后,搅匀;反应液升温至70℃反应,保温反应4小时,反应完成。
[0042]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到242.11g(0.886mol)浅黄色固体状产品。
[0043]
收率93.1%,hplc纯度98.5%,1h nmr(400mhz,d2o):δ4.63(s,1h),3.70
‑
3.94(m,4h),3.32
‑
3.55(m,6h),2.51
‑
3.22(m,6h)。
[0044]
步骤(3):式4化合物2a,4a,8a
‑
三(羧甲基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓一溴三氯盐的制备
[0045]
向5l反应瓶中依次加入1500ml乙腈,和242.11g(0.886mol)式3化合物,搅匀。再在向其中分批加入264.57g(2.8mol)氯乙酸,搅匀。加完后,室温搅拌反应2小时,然后升温至回流反应,保温回流反应8小时,反应结束。
[0046]
反应液冷却至室温,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到468.04g(0.841mol)黄色固体状产品。
[0047]
收率94.9%,hplc纯度99.5%,1h nmr(400mhz,d2o):δ5.71(s,1h),4.22(s,6h),3.72
‑
3.99(m,4h),3.41
‑
3.64(m,6h),2.62
‑
3.29(m,6h)。
[0048]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0049]
向5l反应瓶中加入2000ml水和200g(5.0mol)氢氧化钠,搅拌下再向其中分批加入468.04g(0.841mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温80℃反应6小时,反应结束。
[0050]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入500g 001
×
7树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到265.09g(0.765mol)白色固体状产品。
[0051]
收率91.0%,hplc纯度99.3%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.32(m,4h),3.17(m,12h)。
[0052]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0053]
向5l反应瓶中依次加入1500ml水和265.09g(0.765mol)式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
12,控制温度不超过30℃,搅匀。再向其中加入39.82g(0.3825mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应4小时,反应结束。
[0054]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入适量001
×
7树脂调ph至
4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷丙酮洗涤,抽干,得到328.08g(0.728mol)白色固体状产品。
[0055]
收率95.2%,hplc纯度99.3%,产品ir,ms,元素分析,1h nmr均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0056]
步骤(6):式7化合物钆布醇的制备
[0057]
向2l反应瓶中依次加入1000ml水和328.08g(0.728mol)式6化合物,再向其中加入79.03g(0.218mol)三氧化二钆,搅匀,加热至90℃反应5小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0058]
向滤液中加入280g经活化处理的d301弱碱性阴离子树脂,室温搅拌3小时,过滤,滤除树脂。
[0059]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到396.21g(0.655mol)白色固体状产品。
[0060]
收率90%,六步反应总收率65.6%。
[0061]
产品纯度:99.94%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0062]
实施例2:
[0063][0064]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0065]
保持氮气微正压,向1l反应瓶中依次加入300ml乙二醇二甲醚,146.24g(1.0mol)式1化合物三乙烯四胺和370.5g(2.5mol)原甲酸三乙酯,加完后,搅匀;反应液升温至85℃反应,此时反应液出现回流。反应液保温85℃继续反应7小时,反应完成。
[0066]
反应液冷却析晶得150.44g(0.905mol)白色固体状产品。
[0067]
收率90.5%,hplc纯度99.4%,1h nmr(400mhz,cdcl3):δ6.72(s,2h),3.73(7,4h),3.11
‑
3.24(m,8h)。
[0068]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓溴盐的制备
[0069]
向2l反应瓶中依次加入700ml 2
‑
甲基四氢呋喃,150.44g(0.905mol)式2化合物,243.57g(1.3mol)1,2
‑
二溴乙烷和127.19g(1.2mol)碳酸钠,加完后,搅匀;反应液升温至50℃反应,保温反应7小时,反应完成。
[0070]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷石油醚洗涤,抽干,得到237.33g(0.869mol)浅黄色固体状产品。
[0071]
收率96%,hplc纯度98.7%,1h nmr(400mhz,d2o):δ4.63(s,1h),3.70
‑
3.94(m,4h),3.32
‑
3.55(m,6h),2.51
‑
3.22(m,6h)。
[0072]
步骤(3):式4化合物2a,4a,8a
‑
三(羧甲基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓一溴三碘盐的制备
[0073]
向5l反应瓶中依次加入1000ml nmp,和237.33g(0.869mol)式3化合物,搅匀。再在向其中分批加入520.66g(2.8mol)碘乙酸,搅匀。加完后,室温搅拌反应2小时,然后升温至90℃反应,保温90℃反应6小时,反应结束。
[0074]
反应液冷却至室温,向其中加入适量乙酸乙酯和石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙酸乙酯洗涤,抽干,得到701.94g(0.845mol)黄色固体状产品。
[0075]
收率97.2%,hplc纯度99.1%,1h nmr(400mhz,d2o):δ5.71(s,1h),4.22(s,6h),3.74
‑
3.98(m,4h),3.42
‑
3.63(m,6h),2.61
‑
3.29(m,6h)。
[0076]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0077]
向5l反应瓶中加入2000ml水和300g(5.35mol)氢氧化钾,搅拌下再向其中分批加入701.94g(0.845mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至90℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钾固体。保温90℃反应7小时,反应结束。
[0078]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入70.19g硫酸调ph至4
‑
7,再加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到281.28g(0.812mol)白色固体状产品。
[0079]
收率96.1%,hplc纯度99.3%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.32(m,4h),3.17(m,12h)。
[0080]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0081]
向5l反应瓶中依次加入1200ml水,300ml甲醇和281.28g(0.812mol)式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
12,控制温度不超过30℃,搅匀。再在向其中加入84.54g(0.812mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应8小时,反应结束。
[0082]
反应液冷却至室温,不再经进一步处理,直接应用于下一步反应。
[0083]
步骤(6):式7化合物钆布醇的制备
[0084]
向上一步反应液中加入147.17g(0.406mol)三氧化二钆,搅匀,加热至100℃反应6小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0085]
向滤液中加入400g经活化处理的d941弱碱性阴离子交换树脂,室温搅拌3小时,过
滤,滤除树脂。
[0086]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到454.75g(0.752mol)白色固体状产品。
[0087]
两步收率92.6%,六步反应总收率74.3%。
[0088]
产品纯度:99.92%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0089]
实施例3:
[0090][0091]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0092]
保持氮气微正压,向2l反应瓶中依次加入500ml四氢呋喃,146.24g(1.0mol)式1化合物三乙烯四胺和180.15g(3.0mol)甲酸甲酯,加完后,搅匀;反应液升温至66℃反应,此时反应液出现回流。反应液保温66℃继续反应5小时,反应完成。
[0093]
反应液冷却析晶得152.60g(0.918mol)白色固体状产品。
[0094]
收率91.80%,hplc纯度99.4%,1h nmr(400mhz,cdcl3):δ6.71(s,2h),3.73(7,4h),3.11
‑
3.24(m,8h)。
[0095]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓氯盐的制备
[0096]
向2l反应瓶中依次加入600ml乙腈,152.60g(0.918mol)式2化合物,181.69g(1.836mol)1,2
‑
二氯乙烷和243.89g(1.765mol)碳酸钾,加完后,搅匀;反应液升温至70℃反应,保温反应4小时,反应完成。
[0097]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到194.22g(0.849mol)浅黄色固体状产品。
[0098]
收率92.5%,hplc纯度99.0%,1h nmr(400mhz,d2o):δ4.65(s,1h),3.71
‑
3.93(m,4h),3.32
‑
3.55(m,6h),2.52
‑
3.22(m,6h)。
[0099]
步骤(3):式4化合物2a,4a,8a
‑
三(羧甲基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓四氯三钠盐的制备
[0100]
向5l反应瓶中依次加入1500ml乙腈,和194.22g(0.849mol)式3化合物,搅匀。再在向其中分批加入326.15g(2.8mol)氯乙酸钠,搅匀。加完后,室温搅拌反应2小时,然后升温至回流反应,保温回流反应8小时,反应结束。
[0101]
反应液冷却至室温,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到462.87g(0.801mol)黄色固体状产品。
[0102]
收率94.3%,hplc纯度99.4%,1h nmr(400mhz,d2o):δ5.68(s,1h),4.11(s,6h),3.72
‑
3.97(m,4h),3.40
‑
3.61(m,6h),2.61
‑
3.29(m,6h)。
[0103]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0104]
向5l反应瓶中加入2000ml水和120g(5.0mol)氢氧化锂,搅拌下再向其中分批加入462.87g(0.801mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化锂固体。保温80℃反应6小时,反应结束。
[0105]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入500gamberlite ir=120树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到253.87g(0.733mol)白色固体状产品。
[0106]
收率91.5%,hplc纯度99.2%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.32(m,4h),3.17(m,12h)。
[0107]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0108]
向5l反应瓶中依次加入1500ml水和253.87g(0.733mol)式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
12,控制温度不超过30℃,搅匀。再向其中加入86.54g(0.831mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应4小时,反应结束。
[0109]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入适量001
×
7树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷丙酮洗涤,抽干,得到312.71g(0.694mol)白色固体状产品。
[0110]
收率94.7%,hplc纯度99.4%,产品ir,ms,元素分析,1h nmr均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0111]
步骤(6):式7化合物钆布醇的制备
[0112]
向2l反应瓶中依次加入1000ml水和312.71g(0.694mol)式6化合物,再向其中加入365.89g(1.388mol)三氯化钆,搅匀,加热至90℃反应5小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0113]
向滤液中加入323g经活化处理的a100弱碱阴离子树脂,室温搅拌3小时,过滤,滤除树脂。
[0114]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到396.17g(0.655mol)白色固体状产品。
[0115]
收率94.4%,六步反应总收率65.50%。
[0116]
产品纯度:99.54%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0117]
实施例4:
[0118][0119]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0120]
保持氮气微正压,向2l反应瓶中依次加入900ml叔丁基甲醚,146.24g(1.0mol)式1化合物三乙烯四胺和296.32g(4.0mol)甲酸乙酯,加完后,搅匀;反应液升温至56℃反应,此时反应液出现回流。反应液保温56℃继续反应24小时,反应完成。
[0121]
反应液冷却析晶得154.10g(0.927mol)白色固体状产品。
[0122]
收率92.7%,hplc纯度99.5%,1h nmr(400mhz,cdcl3):δ6.73(s,2h),3.74(7,4h),3.13
‑
3.25(m,8h)。
[0123]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓碘盐的制备
[0124]
向2l反应瓶中依次加入600ml乙腈,154.10g(0.927mol)式2化合物,783.85g(2.781mol)1,2
‑
二碘乙烷和243.89g(1.765mol)碳酸钾,加完后,搅匀;反应液升温至70℃反应,保温反应4小时,反应完成。
[0125]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到277.81g(0.868mol)浅黄色固体状产品。
[0126]
收率93.6%,hplc纯度99.2%,1h nmr(400mhz,d2o):δ4.63(s,1h),3.71
‑
3.96(m,4h),3.30
‑
3.55(m,6h),2.51
‑
3.22(m,6h)。
[0127]
步骤(3):式4化合物2a,4a,8a
‑
三(羧甲基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓三氯一碘三钾盐的制备
[0128]
向5l反应瓶中依次加入1500ml乙腈,和277.81g(0.868mol)式3化合物,搅匀。再在向其中分批加入371.23g(2.8mol)氯乙酸钾,搅匀。加完后,室温搅拌反应2小时,然后升温至回流反应,保温回流反应8小时,反应结束。
[0129]
反应液冷却至室温,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到592.01g(0.825mol)黄色固体状产品。
[0130]
收率95.0%,hplc纯度99.3%,1h nmr(400mhz,d2o):δ5.67(s,1h),4.10(s,6h),3.72
‑
3.97(m,4h),3.40
‑
3.61(m,6h),2.61
‑
3.29(m,6h)。。
[0131]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0132]
向5l反应瓶中加入2000ml水和200g(5.0mol)氢氧化钠,搅拌下再向其中分批加入592.01g(0.825mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温80℃反应6小时,反应结束。
[0133]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入700gdowex50
‑
x8树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到262.90g(0.759mol)白色固体状产品。
[0134]
收率92.0%,hplc纯度99.5%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.33(m,4h),3.17(m,12h)。
[0135]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0136]
向5l反应瓶中依次加入1500ml水和262.90g(0.759mol)白色固体状产品式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
10,控制温度不超过30℃,搅匀。再向其中加入395.10g(3.795mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应4小时,反应结束。
[0137]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入适量001
×
7树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷丙酮洗涤,抽干,得到325.85g(0.723mol)白色固体状产品。
[0138]
收率95.3%,hplc纯度99.5%,产品ir,ms,元素分析,1h nmr均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0139]
步骤(6):式7化合物钆布醇的制备
[0140]
向2l反应瓶中依次加入1000ml水和325.85g(0.723mol)式6化合物,再向其中加入571.77g(2.169mol)三氯化钆,搅匀,加热至90℃反应5小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0141]
向滤液中加入651g经活化处理的d301弱碱性阴离子树脂,室温搅拌3小时,过滤,滤除树脂。
[0142]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到416.33g(0.688mol)白色固体状产品。
[0143]
收率94.7%,六步反应总收率68.4%。
[0144]
产品纯度:99.39%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0145]
实施例5:
[0146][0147]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0148]
保持氮气微正压,向2l反应瓶中依次加入600ml乙二醇二乙醚,146.24g(1.0mol)式1化合物三乙烯四胺和460.3g(10mol)甲酸,加完后,搅匀;反应液升温至120℃反应,此时反应液出现回流。反应液保温120℃继续反应24小时,反应完成。
[0149]
反应液减压蒸馏,蒸除溶剂和大部分剩余甲酸。向剩余物中加入1500ml叔丁基甲醚,搅匀,所得溶液依次用碳酸氢钠溶液,水和饱和食盐水洗涤,并用无水硫酸钠干燥。向干燥后所得到的叔丁基甲醚溶液加入适量石油醚,再冷却析晶得153.43g(0.923mol)白色固体状产品。
[0150]
收率92.3%,hplc纯度99.0%,1h nmr(400mhz,cdcl3):δ6.72(s,2h),3.72(7,4h),3.11
‑
3.24(m,8h)。
[0151]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓甲磺酸盐的制备
[0152]
向5l反应瓶中依次加入1600ml乙腈,153.43g(0.923mol)式2化合物,817.09g(3.744mol)1,2
‑
二甲磺酰氧基乙烷和243.89g(1.765mol)碳酸钾,加完后,搅匀;反应液升温至70℃反应,保温反应4小时,反应完成。
[0153]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到253.38g(0.879mol)浅黄色固体状产品。
[0154]
收率95.2%,hplc纯度98.7%,1h nmr(400mhz,d2o):δ4.66(s,1h),3.72
‑
3.95(m,4h),3.32
‑
3.55(m,6h),2.54
‑
3.22(m,9h)。
[0155]
步骤(3):式4化合物2a,4a,8a
‑
三(叔丁氧羰甲
[0156]
基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓一甲磺酸三溴盐的制备
[0157]
向5l反应瓶中依次加入1500ml乙腈,和253.38g(0.879mol)式3化合物,搅匀。再在
向其中分批加入624.16g(3.2mol)溴乙酸叔丁酯,搅匀。加完后,室温搅拌反应2小时,然后升温至回流反应,保温回流反应8小时,反应结束。
[0158]
反应液冷却至室温,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到724.07g(0.829mol)黄色固体状产品。
[0159]
收率94.3%,hplc纯度99.3%,1h nmr(400mhz,d2o):δ5.68(s,1h),4.21(s,6h),3.74
‑
3.98(m,4h),3.42
‑
3.63(m,6h),2.61
‑
3.29(m,9h),1.35(s,27h)。
[0160]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0161]
向5l反应瓶中加入2000ml水和280g(5.0mol)氢氧化钾,搅拌下再向其中分批加入724.07g(0.829mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至80℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钾固体。保温100℃反应6小时,反应结束。
[0162]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入500g盐酸ph至4
‑
7,室温搅拌16小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到268.48g(0.775mol)白色固体状产品。
[0163]
收率93.5%,hplc纯度99.1%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.32(m,4h),3.17(m,12h)。
[0164]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0165]
向5l反应瓶中依次加入1500ml水和268.48g(0.775mol)式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
12,控制温度不超过30℃,搅匀。再向其中加入96.93g(0.931mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应4小时,反应结束。
[0166]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入适量001
×
7树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷丙酮洗涤,抽干,得到328.18g(0.729mol)白色固体状产品。
[0167]
收率94.0%,hplc纯度99.3%,产品ir,ms,元素分析,1h nmr均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0168]
步骤(6):式7化合物钆布醇的制备
[0169]
向2l反应瓶中依次加入1000ml水和328.51g(0.729mol)式6化合物,再向其中加入960.86g(3.645mol)三氯化钆,搅匀,加热至90℃反应5小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0170]
向滤液中加入985g经活化处理的d301弱碱性阴离子树脂,室温搅拌3小时,过滤,滤除树脂。
[0171]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到418.80g(0.693mol)白色固体状产品。
[0172]
收率95%,六步反应总收率72.8%。
[0173]
产品纯度:98.94%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0174]
实施例6:
[0175][0176]
步骤(1):式2化合物1,2
‑
双(4,5
‑
二氢
‑
1h
‑
咪唑
‑1‑
基)乙烷的制备
[0177]
保持氮气微正压,向2l反应瓶中依次加入600ml乙二醇二甲醚,146.24g(1.0mol)式1化合物三乙烯四胺和357.48g(3.0mol)dmf
‑
dma,加完后,搅匀;反应液升温至85℃反应,此时反应液出现回流。反应液保温85℃继续反应5小时,反应完成。
[0178]
反应液冷却析晶得157.59g(0.948mol)白色固体状产品。
[0179]
收率94.8%,hplc纯度99.5%,1h nmr(400mhz,cdcl3):δ6.72(s,2h),3.72(m,4h),3.10
‑
3.25(m,8h)。
[0180]
步骤(2):式3化合物1,2,2a1,3,4,6,7,8
‑
八氢
‑
5h
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
6a
‑
鎓对甲苯磺酸盐的制备
[0181]
向2l反应瓶中依次加入600ml乙腈,157.59g(0.948mol)式2化合物,361.54g(0.976mol)1,2
‑
二对甲苯磺酰氧基乙烷和243.89g(1.765mol)碳酸钾,加完后,搅匀;反应液升温至70℃反应,保温反应4小时,反应完成。
[0182]
反应液趁热过滤,滤液自然冷却,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,析出大量浅黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到322.37g(0.885mol)浅黄色固体状产品。
[0183]
收率93.5%,hplc纯度99.4%,1h nmr(400mhz,d2o):δ7.74(d,2h),7.45(d,2h),4.66(s,1h),3.70
‑
3.94(m,4h),3.32
‑
3.55(m,6h),2.59
‑
3.22(m,6h),2.45(s,3h)。
[0184]
步骤(3):式4化合物2a,4a,8a
‑
三(乙氧羰甲
[0185]
基)
‑
1,2,2a,2a1,3,4,4a,5,6,7,8,8a
‑
十二氢
‑
2a,4a,6a,8a
‑
四氮环戊[fg]苊烯
‑
2a,4a,6a,8a
‑
四鎓一对甲苯磺酸三氯盐的制备
[0186]
向5l反应瓶中依次加入1500ml乙腈,和322.37g(0.885mol)式3化合物,搅匀。再在向其中分批加入343.14g(2.8mol)氯乙酸乙酯,搅匀。加完后,室温搅拌反应2小时,然后升温至回流反应,保温回流反应8小时,反应结束。
[0187]
反应液冷却至室温,向其中加入适量石油醚,冷却至0℃进行析晶。保温析晶过夜,
析出大量黄色固体。过滤,收集滤饼,滤饼用适量冷乙腈洗涤,抽干,得到616.82g(0.843mol)黄色固体状产品。
[0188]
收率95.2%,hplc纯度99.4%,1h nmr(400mhz,d2o):δ7.74(d,2h),7.45(d,2h),5.69(s,1h),4.22(s,6h),4.15(q,6h),3.74
‑
3.98(m,4h),3.42
‑
3.63(m,6h),2.61
‑
3.29(m,6h),2.46(s,3h),1.20(t,9h)。
[0189]
步骤(4):式5化合物1,4,7,10
‑
四氮杂环十二烷
‑
1,4,7
‑
三乙酸的制备
[0190]
向5l反应瓶中加入2000ml水和200g(5.0mol)氢氧化钠,搅拌下再向其中分批加入616.82g(0.843mol)式4化合物,控制温度不超过30℃,搅匀。加完后,升温至100℃反应,反应过程中监控体系ph值,使其保持在9
‑
14,如果ph低于9,则需向其中补加氢氧化钠固体。保温100℃反应6小时,反应结束。
[0191]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入500g diaion sk
‑
1b树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷甲醇洗涤,抽干,得到270.68g(0.781mol)白色固体状产品。
[0192]
收率92.7%,hplc纯度99.5%,1h nmr(400mhz,d2o):δ3.54(s,6h),3.32(m,4h),3.17(m,12h)。
[0193]
步骤(5):式6化合物2,2',2
”‑
(10
‑
((2r,3s)
‑
1,3,4
‑
三羟基丁
‑2‑
基)
‑
1,4,7,10
‑
四氮杂环十二烷)
‑
1,4,7
‑
三乙酸的制备
[0194]
向5l反应瓶中依次加入1500ml水和270.68g(0.781mol)式5化合物,搅匀,再向其中滴加适量饱和氢氧化钠溶液,调体系ph值8
‑
12,控制温度不超过30℃,搅匀。再向其中加入86.54g(0.831mol)2,3
‑
环氧基
‑
1,4
‑
丁二醇,搅匀。加完后,室温搅拌反应2小时,然后升温至60℃反应,保温反应4小时,反应结束。
[0195]
反应液旋蒸,除去适量水。剩余液冷却至室温,向其中加入适量001
×
7树脂调ph至4
‑
7,室温搅拌1小时,过滤,收集滤液。向滤液中加入适量氯化钠,搅匀。反应液冷却至
‑
5℃,冷却析晶过夜。析出大量白色固体。过滤,收集滤饼,滤饼用适量冷丙酮洗涤,抽干,得到330.02g(0.733mol)白色固体状产品。
[0196]
收率93.8%,hplc纯度99.4%,产品ir,ms,元素分析,1h nmr均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0197]
步骤(6):式7化合物钆布醇的制备
[0198]
向2l反应瓶中依次加入1000ml水和330.02g(0.733mol)式6化合物,再向其中加入193.23g(0.733mol)三氯化钆,搅匀,加热至90℃反应5小时,反应结束。反应液冷却至室温,用0.2微米滤膜过滤,滤除不溶物。
[0199]
向滤液中加入1650g经活化处理的a100弱碱阴离子树脂,室温搅拌3小时,过滤,滤除树脂。
[0200]
滤液旋干,剩余物用适量乙醇打浆1小时,再用乙醇
‑
水(8:1,v/v)重结晶,得到414.45g(0.685mol)白色固体状产品。
[0201]
收率93.5%,六步反应总收率68.6%。
[0202]
产品纯度:99.78%(hplc);产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0203]
对比例1:
[0204]
根据常规技术中公开的方案1(inorg.chem.1997,36,6086
‑
6093中scheme 1合成方案)以轮环藤宁为起始原料来制备钆布醇。
[0205]
总收率:37.9%,产品纯度:95.54%(hplc)。产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0206]
对比例2:
[0207]
根据常规技术中公开的方案2(inorg.chem.1997,36,6086
‑
6093中scheme 2合成方案)以轮环藤宁为起始原料来制备钆布醇。
[0208]
总收率:35.5%,产品纯度:94.38%(hplc)。产品ir,ms,元素分析均与文献(inorg.chem.1997,36,6086
‑
6093)报道数据一致。
[0209]
综合实施例1
‑
6和对比例1
‑
2的总收率和产品纯度结果对比分析
[0210]
类型实施例1实施例2实施例3实施例4实施例5实施例6对比例1对比例2总收率65.6%74.3%65.5%68.4%72.8%68.6%37.9%35.5%纯度99.94%99.92%99.54%99.39%98.94%99.78%95.54%94.38%
[0211]
本专利的合成方法具有以下优点:
[0212]
1、原料价格便宜,主料三乙烯四胺每公斤单价130元,仅为轮环藤宁价格的3%;
[0213]
2、总收率高,本专利总收率65.5%
‑
74.3%远高于文献报道工艺路线最高收率46%;
[0214]
3、只引入了一次保护基,少于文文献报道工艺路线中的两次保护基,本专利的原子经济性更高,操作也更加便捷;
[0215]
4、产品纯度高,所得产品纯度最高高达99.94%,直接满足临床应用的要求。
[0216]
综上,本方法与现行文献报道方法相比,优势非常明显。
[0217]
尽管以上对本发明的实施方案进行了描述,但本发明并不局限于上述的具体实施方案和应用领域,上述的具体实施方案仅仅是示意性的、指导性的,而不是限制性的。本领域的普通技术人员在本说明书的启示下和在不脱离本发明权利要求所保护的范围的情况下,还可以做出很多种的形式,这些均属于本发明保护之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1