基于连接酶链式反应和基因编辑技术检测核酸标志物的方法
1.本发明涉及分子诊断检测技术领域,具体地说是涉及一种基于连接酶链式反应和基因编辑技术检测核酸标志物的方法。
背景技术:
2.随着化学、分子生物学及生物医学等学科的发展与交叉,研究检测核酸分子标志物对在基因水平上诊断和治疗重大疾病以及研究不同个体之间的药物响应等具有重要的应用价值。研究表明,许多疾病如新冠肺炎、遗传疾病和癌症的发生及发展都与核酸分子标志物(如基因突变、rna及mircorna(mirna)等)的异常表达密切相关。因此,在生命体系发生变化的初始阶段实现基因突变或rna等核酸标志物的检测对于疾病的早期诊断、个体化治疗和疗效判断等具有重要意义。开发和建立简便、均相、超灵敏度精准检测核酸标志物的新方法必将为疾病的早期诊断和治疗提供强有力的支持。
3.针对核酸分子标志物检测,已报道了多种方法。由于生物样品量少,且其中的核酸分子标志物的含量低,因此,高灵敏度核酸检测一般都需要对目标序列进行预扩增。当前,应用较为广泛的核酸扩增技术主要有聚合酶链式反应(pcr)、环介导等温扩增(lamp)、滚环扩增(rca)等。其中,pcr是当前应用最广的扩增技术。pcr扩增的灵敏度高,但其对单碱基的识别能力差,在大量其它核酸分子存在下,扩增低丰度目标核酸分子容易出现错误的结果。而且常用的实时定量pcr技术需结合taqman探针进行检测,每一个突变位点需要设计与序列相关的taqman探针,费用较高。数字pcr(ddpcr)作为最新一代pcr技术,尽管其灵敏度高、特异性好,但其仪器设备尚昂贵,操作繁琐,这些限制了pcr技术的进一步发展。
4.lcr是一种以连接反应为基础,依靠模板循环扩增的技术。lcr利用两对寡核苷酸探针,每对包含2条dna探针,通过特异性dna连接酶将与靶核酸互补的相邻探针进行连接,连接好的探针在之后的热循环中作为模板,产生指数扩增。由于酶连接反应对单碱基错配的识别能力大大强于引物延伸扩增反应(如pcr及lamp等)的识别能力,因此lcr相比于依靠引物延伸的扩增技术,对单碱基基因突变的检测有更好的特异性。而且lcr仅需要一种连接酶,简单快速,灵敏度与pcr相当。但是,由于lcr独特的反应机理,即反应前、后都是双链dna,仅在长短上存在差异,lcr不像pcr那样可使用taqman探针或分子信标探针等实现均相荧光检测。因此,lcr检测一般需要结合磁珠分离或者电泳分离,操作繁琐,其应用受到限制。虽然在前期研究中,我们课题组及其他课题组基于阳离子共轭聚合物(ccp)荧光共振能量转移技术(fret)及荧光染料实现了lcr的均相检测,但方法灵敏度较低(1fmol/l)。因此,进一步创新发展lcr的均相、高灵敏度检测的新方法面临着重大挑战。
技术实现要素:
5.本发明的目的是提供一种基于连接酶链式反应和基因编辑技术检测核酸标志物的方法。以解决现有方法操作繁琐、灵敏度较低的问题。
6.本发明的目的是这样实现的:一种基于连接酶链式反应和基因编辑技术检测核酸
标志物的方法,包括以下步骤:
7.(a)根据目标核酸标志物的序列,设计两对用于连接酶链式反应的dna探针。其中,第一对dna探针与目标核酸标志物互补,第一对dna探针连接点处的5
′
端进行磷酸化修饰;第二对dna探针与第一对dna探针序列互补或第二对dna探针连接点处的3
′
端突出一个碱基,5
′
端进行磷酸化修饰;在dna探针远离连接点侧的5
′
端设计一处pam序列;
8.(b)将待检测核酸与两对dna探针混合,并加入连接酶进行连接酶链式反应,若目标核酸标志物存在,则反应生成含有pam序列的双链dna连接产物;
9.(c)在步骤(b)所得的连接产物中加入grna、crispr/cas12系统以及报告探针,所述报告探针一端修饰有荧光基团,另一端修饰有猝灭基团;所述grna能够结合双链dna连接产物,并激活crispr/cas12系统,crispr/cas12系统被激活后切割报告探针并释放荧光信号;
10.(d)检测反应系统荧光信号,得到待检测核酸中目标核酸标志物的含量。
11.步骤(a)中,所述pam序列与所述连接点之间间隔10-16个碱基。
12.步骤(a)中,所述目标核酸标志物为dna或rna,当目标核酸标志物为单碱基突变dna时,第二对dna探针连接点处的3
′
端突出碱基在突变碱基的位置。
13.步骤(a)中,rna目标核酸标志物包括microrna、信使rna、环状rna、长链非编码rna、病毒rna。
14.步骤(b)中,连接酶链式反应所用连接酶包括amp dna连接酶、taq dna连接酶、9
°
n dna连接酶;进行连接酶链式反应时,连接温度为45~65℃,热循环次数为20~50,连接酶用量为0.2~20u。
15.步骤(b)中,当目标核酸标志物为rna时,先以rna为模板在连接酶的作用下连接第一对dna探针,所述连接酶为splintr连接酶或t4 rna连接酶2。
16.步骤(c)中,所述grna包含根据目标核酸标志物设计的检测识别区。
17.步骤(c)中,所述crispr/cas12系统的cas12酶可为ascas12a、lbcas12a、fncas12a、arcas12a、lpcas12a、bscas12a、hkcas12a、prcas12a、pxcas12a中的至少一种,此时,pam序列为5
′‑
tttn或5
′‑
ttn,其中n代表任意碱基;所述crispr/cas12系统的cas12酶也可为hkcas12a,此时,pam序列为5
′‑
yyn,其中y代表c或者t碱基,n代表任意碱基;所述crispr/cas12系统的cas12酶也可为cas12b,此时,pam序列为5
′‑
ttn,其中n代表任意碱基。
18.在进行步骤(c)操作之前,可将含pam的探针及其互补探针、grna和crispr/cas12系统混合,检测系统荧光,若有荧光信号,则将步骤(b)所得的连接产物进行lambda核酸外切酶消化后再进行步骤(c)的反应。
19.步骤(c)中,报告探针的荧光基团为fam、fitc、hex、tet、tamra、rox或cy5,猝灭基团为bhq1、tamra、bhq2、bhq3或dabcyl。
20.步骤(d)中,荧光检测可使用荧光检测装置,包括荧光光度计、实时荧光pcr仪酶标仪或荧光检测仪。也可以在蓝光、紫外灯或荧光显微镜下用肉眼直接观察。
21.本发明结合lcr反应特异高效的扩增能力和cas12a独特的核酸内切酶活性,构建了超灵敏级联信号放大体系,提供了一种核酸标志物均相、超灵敏检测的新方法。本发明创新设计探针序列,当lcr扩增产物生成后,cas12a核酸内切酶活性被激活,对两端分别标记荧光及猝灭基团的信号探针进行循环切割,实现目标核酸分子的二次信号放大。直接检测
荧光信号即可对核酸标志物进行定量检测。此外,由于报告探针的序列与目标序列无关,以上反应体系的信号探针还可以推广到任何目标序列,节约了成本、简化了设计。
22.本发明方法将大大拓展lcr技术的应用,既可用于dna检测,也可用于rna检测,为核酸标志物的高灵敏度分析提供均相检测新策略,可为临床诊疗及生物医学研究提供新技术,在重大疾病的早期诊断、个性化用药及突发疫情的有效防控等方面具有重要意义与潜在的应用前景。
附图说明
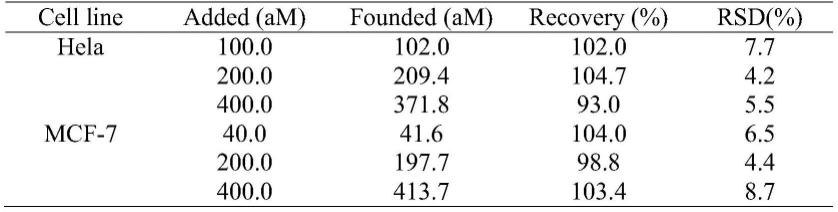
23.图1是基于连接酶链式反应和基因编辑技术检测基因突变的原理示意图。
24.图2是不同浓度的突变型dna扩增产物检测的荧光光谱图。
25.图3是荧光强度随着突变型dna浓度变化的标准曲线图。
26.图4是突变型dna和野生型dna按不同比例混合扩增后的荧光趋势图。
27.图5是实施例2中相同浓度的let-7a、mir-214、mir-141、mir-221和mir-222所对应的荧光信号图。
28.图6是实施例2中不同浓度的mirna-221所产生的实时荧光曲线图。
29.图7是实施例2中荧光强度随着mirna-221浓度变化的标准曲线图。
具体实施方式
30.下面结合实施例对本发明做进一步的阐述,下述实施例仅作为说明,并不以任何方式限制本发明的保护范围。
31.在下述实施例中未详细描述的过程和方法是本领域公知的常规方法,实施例中所用试剂均为分析纯或化学纯,且均可市购或通过本领域普通技术人员熟知的方法制备。下述实施例均实现了本发明的目的。
32.实施例1
33.基于连接酶链式反应和基因编辑技术检测基因突变,检测原理如图1所示,具体检测方法如下:
34.1.选择jak2基因片段为待测位点,野生型序列为5
′‑
cacaagcatttggttttaaattatggagtatgtgtctgtggagacgagagtaagtaa-3
′
,其互补序列为5
′‑
ttacttactctcgtctccacagacacatactccataatttaaaaccaaatgcttgtgtg-3
′
。含有jak2v617f基因突变位点的dna序列为5
′‑
cacaagcatttggttttaaattatggagtatgtttctgtggagacgagagtaagtaa-3
′
,其互补序列为5
′‑
ttacttactctcgtctccacagaaacatactccataatttaaaaccaaatgcttgtgtg-3
′
。设计合成四条lcr探针x、xx、y和yy,其中x探针的碱基序列为5
′‑
po
4-acatactccataatttaaaaccttt-3
′
,xx探针的碱基序列为5
′‑
ttaggttttaaattatggagtatgtt-3
′
,y探针的碱基序列为5
′‑
caagcatctttactcgtctccacagaa-3
′
,yy探针的碱基序列为5
′‑
po
4-tctgtggagacgagtaaagatgcttgt-3
′
,grna碱基序列为5
′‑
uaauuucuacuaaguguagaucucgucuccacagaaaca-3
′
(其中画下划线部分为检测识别区),报告探针的碱基序列为5
′‑
fam-cacacctcagcg-bhq1-3
′
。
35.2.在200μl离心管中加入13.4μl灭菌水、2μm x、y、xx和yy探针水溶液各0.8μl、2.0μl 10
×
amp dna连接酶反应缓冲液(200mm tris-hcl、250mm kcl、100mm mgcl2、5mm腺嘌呤
核苷三磷酸和0.1%曲拉通100,ph 8.3,25℃)、1.0μl不同浓度的突变型双链dna水溶液,然后置于pcr仪中,95℃变性5min,25℃杂交25min,用移液器充分混匀,作为溶液a。在200μl离心管中加入0.4μl 5u/μl amp dna连接酶,从实验开始至加酶结束,整个过程全部在冰上操作,以保持低温环境。将pcr仪首先预加热至95℃,加完酶后立即将溶液a转移至预加热好的pcr仪中,95℃保持3min,使四条探针和突变型dna目标链充分变性。然后95℃保持30s再降温至60℃保持30s,按照这个温度条件进行34个热循环,扩增结束后立即将连接产物放在冰上。
36.3.在步骤2的lcr产物中加入0.3μl 5u/μl lambda核酸外切酶,37℃恒温孵育1.5h,然后80℃保持30min使lambda核酸外切酶失去活性。该酶能够作用于双链dna,沿5
′→3′
方向逐步切去5
′
端单核苷酸。最合适的底物是5
′
端磷酸化的双链dna,加入lambda核酸外切酶后会将步骤2中未参与反应的连接探针进行消化以降低背景信号。
37.4.在200μl离心管中加入1.0μl 10
×
nebuffer 2.1(50mm nacl、10mm tris-hcl、10mm mgcl2、100μg/ml牛血清白蛋白,ph 7.9,25℃)。然后加入1.0μl 400nm报告探针、0.5μl 100mm二硫苏糖醇、0.5μl 40u/μl重组rna酶抑制剂、1.0μl 2μm grna溶液和0.5μl rnase-free水作为溶液b。取5.0μl步骤3中的扩增产物加入到溶液b中,然后加入0.5μl 1μm cas12a核酸酶,充分混匀后加入至溶液b中,整个过程全部在冰上操作。然后将盛有溶液b的离心管转移至pcr仪中,37℃恒温孵育60min,然后80℃保持20min使cas12a酶失去活性。反应结束后,在200μl离心管中加入90μl 1
×
te缓冲溶液(10mm tris-hcl,1mm edta,ph 8.0,25℃)充分稀释混匀,然后将稀释好的样品全部转移至200μl微量荧光池中,在f-7000荧光光谱仪上进行光谱扫描。激发波长为480nm,发射波长扫描范围为500nm-650nm,在525nm发射波长处记录荧光值。
38.5.在优化好的实验条件下,利用该方法对突变型dna浓度进行定量检测。图2是不同浓度的突变型dna和不加任何目标物的空白实验所产生的荧光光谱图,由图可以看出,随着突变型dna浓度由0.1fm增加至1pm,525nm发射波长处所对应的荧光值呈现明显的上升趋势。平行进行三组扩增实验,并对不同浓度的突变型dna进行检测,记录各个浓度的突变型dna在525nm发射波长处的荧光值。图3是三组平行实验的定量标准曲线,线性方程为
△
f=135.9lg(c/fm)+177.0,线性相关系数r=0.9948。由图3可以看出,突变型dna浓度在0.1fm-1pm浓度范围内,荧光强度与突变型d na浓度的对数呈现良好的线性关系。检出限(3σ,n=11)经计算为0.077fm。对100fm突变型dna平行测定7次计算精密度,其相对标准偏差为4.6%。
39.6.等位基因的突变频率与某些疾病密切相关,对患者进行等位基因突变频率检测可以达到诊断的目的。因此,对等位基因突变频率进行测定具有非常重要的意义。利用该实验方法对等位基因突变频率进行测定,其中突变型dna和野生型dna按不同比例进行混合(0%、0.5%、1%、2%、5%、10%、20%、50%、100%),二者浓度之和为1pm,实验步骤同上。图4显示的是不同突变频率下所对应的荧光值,从图中可以明显看出随着突变频率(即突变型dna的占比)的增加,其所对应的荧光强度呈现明显的上升趋势。该实验方法能够准确检测到低至0.5%的突变频率,具有较高的特异性。
40.实施例2
41.连接酶链式反应结合基因编辑技术在检测microrna(mirna)中的应用,具体检测
方法如下:
42.1、根据mir-221的rna序列5
′‑
agcuacauugucugcuggguuuc-3
′
设计合成了四条lcr探针,x1、x2、y1和y2,其中探针x1(碱基序列为5
′‑
caagcatctttcgaaacccagca-3
′
)与探针y1(碱基序列为5
′‑
po
4-gacaatgtagctctcaac-3
′
)分别与目标序列mir-221部分互补。探针x2(碱基序列为5
′‑
po
4-tgctgggtttcgaaagatgcttgt-3
′
)与探针x1互补,探针y2(碱基序列为5
′‑
gttgagagctacattgtc-3
′
)与探针y1互补。mir-grna碱基序列为5
′‑
uaauuucuacuaaguguagaugaaacccagcagacaaugu-3
′
(其中画下划线为检测识别区)。
43.2、在200μl离心管中加入5.3μl rnase-free水、1.0μl 10
×
splintr连接酶反应缓冲液(500mm tris-hcl、100mm mgcl2、10mm腺嘌呤核苷三磷酸、100mm二硫苏糖醇,ph 7.5,25℃)、100nm探针x1、y1各1.0μl、0.5μl 40u/μl重组核糖核酸酶抑制剂和1.0μl不同浓度的mirna-221水溶液(同时做空白实验,空白实验中加1.0μlrnase-free水),充分混合均匀,作为溶液a1(整个过程全部在冰上进行操作)。将盛有溶液a1的离心管置于pcr仪中,80℃加热3min,使得探针x1、y1和mir-221变性,然后降至25℃恒温保持20min,使得探针x1和探针y1与目标物mir-221充分杂交。20min后于溶液a1中加入0.2μl 25u/μl splintr连接酶,25℃恒温孵育40min,然后将溶液a1加热至80℃保持20min,使splintr连接酶充分变性失活,反应结束后立即将溶液a1置于冰上保存。
44.3、取一个新的200μl离心管,向管中加入相应的lcr反应混合液,其中lcr反应混合液包括13.6μl rnase-free水、2.0μl 10
×
amp dna连接酶反应缓冲液(200mm tris-hcl、250mm kcl、100mm mgcl2、5mm腺嘌呤核苷三磷酸、0.1%曲拉通100,ph 8.3,25℃)、1μm四条lcr探针x1、x2、y1和y2各0.5μl,2.0μl步骤2中连接产物和0.4μl 5u/μl amp dna连接酶,充分混匀作为溶液b1,整个过程全部在冰上操作,以保持低温环境。将pcr仪首先预加热至95℃,加酶结束后立即将溶液b1转移至预加热好的pcr仪中,95℃保持3min,使四条探针和步骤2中的连接产物变性,然后进行38个热循环,每个热循环95℃30s、58℃30s,反应结束后立即将扩增产物放在冰上保存。
45.4、在200μl离心管中加入3.5μl rnase-free水、1.0ul 10
×
nebuffer 2.1(50mm nacl、10mm tris-hcl、10mm mgcl2、100μg/ml牛血清白蛋白,ph 7.9,25℃)、1.0μl 400nm报告探针、0.5μl 100mm二硫苏糖醇、0.5μl 40u/μl重组核糖核酸酶抑制剂、1.0μl 2μm mir-grna水溶液,充分混匀后作为溶液c。取2.0μl步骤3中的扩增产物加入到溶液c中,然后立即加入0.5μl 1μm cas12a核酸酶,充分混匀后转移至荧光定量pcr仪中,在37℃下进行40个热循环,每2min采集一次荧光强度信号。
46.5、对于mirna检测,由于mirna的序列较短,并且同一族的mirna序列有时极为相近,因此要对其进行特异性检测实验。图5显示的是相同浓度(4fm)的let-7a、mir-214、mir-141、mir-222和mir-221所对应的荧光信号差异图。其中let-7a、mir-214、mir-141引起的荧光信号增长极低,只有mir-222所产生的信号值略微偏高,这是因为其与mir-221存在部分相同的碱基序列,尽管如此,mir-221与mir-222之间的信号值也具有显著的差异,表明该方法对于mir-221的检测具有良好的特异性。
47.6、在已优化好的实验基础上,对该方法的灵敏度进行测定。图6是不同浓度的mir-221及空白实验所产生的实时荧光曲线,从图中可以看出当目标物浓度为0.4am时仍然与空白之间存在明显的差别。图7是荧光强度随着mir-221浓度变化的标准曲线图,在图6的基础
上取反应时间为30min时所对应的荧光强度与mir-221浓度的对数作图得到标准曲线。在图7中可以看到,目标物浓度在0.4am-40am范围内存在一个线性关系,其线性方程为
△
f1=8019.9lg(c/am)+13809.8,线性相关系数r1=0.997。当目标物浓度范围在40am
–
4fm之间存在另外一个线性关系,其线性方程为
△
f2=94226.5lg(c/am)-124069.2,线性相关系数r2=0.993。由此可知,两段线性均存在良好的线性关系。通过平行测定7次200am mir-221得出该方法的精密度,其相对标准偏差rsd为2.9%。
48.7、为了评价这一方法在实际样品中的适用性,我们测定了宫颈癌细胞(hela)与乳腺癌细胞(mcf-7)中mir-221的含量。经测定,100pg hela和mcf-7细胞提取样品中分别含有114和24.5zmol的mir-221。然后我们进行加标回收率测定,在hela和mcf-7细胞提取的总小rna中加入不同量的mir-221。回收率为93.0
–
104.7%,相应的rsd为4.4-8.7%(表1),表明这一方法能够用于细胞中提取样品的测定。
49.表1.hela和mcf-7细胞提取样品中的mir-221的回收率测定(n=3).
50.
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1