SHP2与CDK4/6双靶点抑制化合物合成及其制备方法与应用
本发明属于药物化学领域,具体涉及一种含shp2与cdk4/6双靶点抑制剂合成及其制备方法与应用。
背景技术:
1、shp2是一个在体内广泛存在的非受体型蛋白酪氨酸磷酸酶,具有两个n末端src同源性2结构域(n-sh2和c-sh2)、催化结构域(ptp)和c末端尾部。这两个sh2结构域控制shp2的亚细胞定位和功能调节。作为血小板源性生长因子(pdgf)、表皮生长因子(egf)、成纤维细胞因子(fgf)、白细胞介素-3(il-3)、白血病抑制因子(lif)及α-干扰素(inf-α)等生长因子的下游信号分子,shp2参与ras/mark通路、pi3k/akt通路、jak/stat通路、jnk通路等在内的多条信号通路。研究显示,shp2突变或过度活化将会导致努南综合征、青少年骨髓单核细胞白血病、骨髓增生异常综合征、b细胞急性淋巴细胞白血病及固体瘤(肺癌、结肠癌、神经母细胞瘤、黑色素瘤、肝癌)的发生。
2、cdk是一类丝氨酸/苏氨酸蛋白激酶。其中cdk4/6是细胞周期的核心调控者,调控着细胞周期各阶段的转变,是抗肿瘤药物的重要作用靶点之一。cdk4/6抑制剂可通过抑制肿瘤细胞周期从g1期向s期转变、激活抗肿瘤免疫应答和影响肿瘤微环境等机制产生抗肿瘤活性,临床上对某些类型的肿瘤有较好的疗效。但长时间的cdk4/6抑制剂治疗,极易出现肿瘤耐药的现象,导致抑制剂失去疗效。
3、研究发现,shp2抑制剂与cdk4/6抑制剂联合给药具有较强的协同作用。目前该联合给药已进入临床i期,用于治疗头颈癌、非小细胞肺癌、结肠癌等恶性肿瘤。
4、本发明通过药效团融合和合理药物设计,制备出一类全新shp2与cdk4/6双靶点抑制剂,为后期解决激酶和磷酸酶耐药问题提供基础。
技术实现思路
1、本发明需要解决的技术问题之一是shp2抑制剂和cdk4/6抑制剂面临的临床疗效差、长期使用易出现耐药问题。因此提供一种新型含shp2与cdk4/6双靶点抑制化合物,为后续药物开发提供可能。
2、解决上述技术问题的方案如下:
3、1、—种如通式i所示的化合物,及其药学上可接受的盐、对映异构体、非对映异构体、互变异构体、溶剂化物、多晶型物或前药
4、
5、环a选自c3-c6环烷基、c6-c10芳环、c6-c10杂芳环、单杂环、螺环、桥环、并环;其中所述环烷基、芳环、杂芳环、单杂环、螺环、桥环、并环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的环烷基、芳环、杂芳环、碳环、单杂环、螺环、桥环、并环可任选地被一个或多个取代基取代;
6、环b选自芳环、杂芳环、碳环、杂环;其中所述芳环、杂芳环、碳环、杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的芳环、杂芳环、碳环、杂环可任选地被一个或多个取代基取代;
7、l1为化学键、nra、c1-c2的碳链、o、s(o)m,其中m选自0、1和2;
8、l2为化学键、nra、c1-c2的碳链、o、s(o)m,其中m选自0、1和2;
9、y1为n或cr3,其中r3选自氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c1-c6氘代烷基、c1-c6烷氧基、c2-c6烯基、c2-c6炔基或c3-c6环烷基;
10、y2为n或cr4,其中r4选自氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c1-c6氘代烷基、c1-c6烷氧基、c2-c6烯基、c2-c6炔基或c3-c6环烷基;
11、r1、r2分别为氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
12、或者,r1和r2与它们都相连的碳原子一起形成3至12元单杂环或多环杂环,其中所述单杂环或多环杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的单杂环或多环杂环可以是未取代的或被一个或多个取代基取代;
13、或者,r1和r2与它们都相连的碳原子一起形成
14、
15、其中,w不存在或者选自cr6r7、o、nrb、s(o)m;其中m选自0、1和2;
16、c环不存在或者选自c3-c7元单环或c5-c12元多环;
17、ra、rb各自独立选自氢、氘原子、c1-c6烷基、c3-c6环烷基、c6-c10芳基,c6-c10杂芳基;
18、r4、r5、r6、r7各自独立选自氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
19、或者r4和r5与它们都相连的碳原子一起形成co、c=nh、c3-c12元杂环或c3-c8环烷基;
20、2、—种如通式ii所示的化合物,及其药学上可接受的盐、对映异构体、非对映异构体、互变异构体、溶剂化物、多晶型物或前药
21、
22、环a选自c3-c6环烷基、c6-c10芳环、c6-c10杂芳环、单杂环、螺环、桥环、并环;其中所述环烷基、芳环、杂芳环、单杂环、螺环、桥环、并环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的环烷基、芳环、杂芳环、碳环、单杂环、螺环、桥环、并环可任选地被一个或多个取代基取代;
23、环b选自芳环、杂芳环、碳环、杂环;其中所述芳环、杂芳环、碳环、杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的芳环、杂芳环、碳环、杂环可任选地被一个或多个取代基取代;
24、l1为化学键、nra、c1-c2的碳链、o、s(o)m,其中m选自0、1和2;
25、l2为化学键、nra、c1-c2的碳链、o、s(o)m,其中m选自0、1和2;
26、r1、r2分别为氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
27、或者,r1和r2与它们都相连的碳原子一起形成3至12元单杂环或多环杂环,其中所述单杂环或多环杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的单杂环或多环杂环可以是未取代的或被一个或多个取代基取代;
28、或者,r1和r2与它们都相连的碳原子一起形成
29、
30、其中,w不存在或者选自cr6r7、o、nrb、s(o)m;其中m选自0、1和2;
31、c环不存在或者选自c3-c7元单环或c5-c12元多环;
32、ra、rb各自独立选自氢、氘原子、c1-c6烷基、c3-c6环烷基、c6-c10芳基,c6-c10杂芳基;
33、r4、r5、r6、r7各自独立选自氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
34、或者r4和r5与它们都相连的碳原子一起形成co、c=nh、c3-c12元杂环或c3-c8环烷基;
35、3、—种如通式iii所示的化合物,及其药学上可接受的盐、对映异构体、非对映异构体、互变异构体、溶剂化物、多晶型物或前药
36、
37、a环选自氢、
38、环b选自芳环、杂芳环、碳环、杂环;其中所述芳环、杂芳环、碳环、杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的芳环、杂芳环、碳环、杂环可任选地被一个或多个取代基取代;
39、l2为化学键、nra、c1-c2的碳链、o、s(o)m,其中m选自0、1和2;
40、r1、r2分别为氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
41、或者,r1和r2与它们都相连的碳原子一起形成3至12元单杂环或多环杂环,其中所述单杂环或多环杂环可任选地含有选自n、o和s(o)m的杂原子;其中m选自0、1和2;其中所述的单杂环或多环杂环可以是未取代的或被一个或多个取代基取代;
42、或者,r1和r2与它们都相连的碳原子一起形成
43、
44、其中,w不存在或者选自cr6r7、o、nrb、s(o)m;其中m选自0、1和2;
45、c环不存在或者选自c3-c7元单环或c5-c12元多环;
46、ra、rb各自独立选自氢、氘原子、c1-c6烷基、c3-c6环烷基、c6-c10芳基,c6-c10杂芳基;
47、r4、r5、r6、r7各自独立选自氢、氘原子、卤素、氨基、羟基、氰基、硝基、羧基、磺酸基、c1-c6烷基、c3-c6环烷基、c6-c10芳基、c6-c10杂芳基;其中所述的氨基、羟基、芳基、杂芳基、环烷基可任选地被一个或多个取代基取代;
48、或者r4和r5与它们都相连的碳原子一起形成co、c=nh、c3-c12元杂环或c3-c8环烷基;
49、一种药物组合物,其特征在于含有化合物i-iii和药学可接受的辅料。
50、所的药物组合物,其特征在于药物组合物制成片剂、胶囊剂、注射液或冻干粉剂。
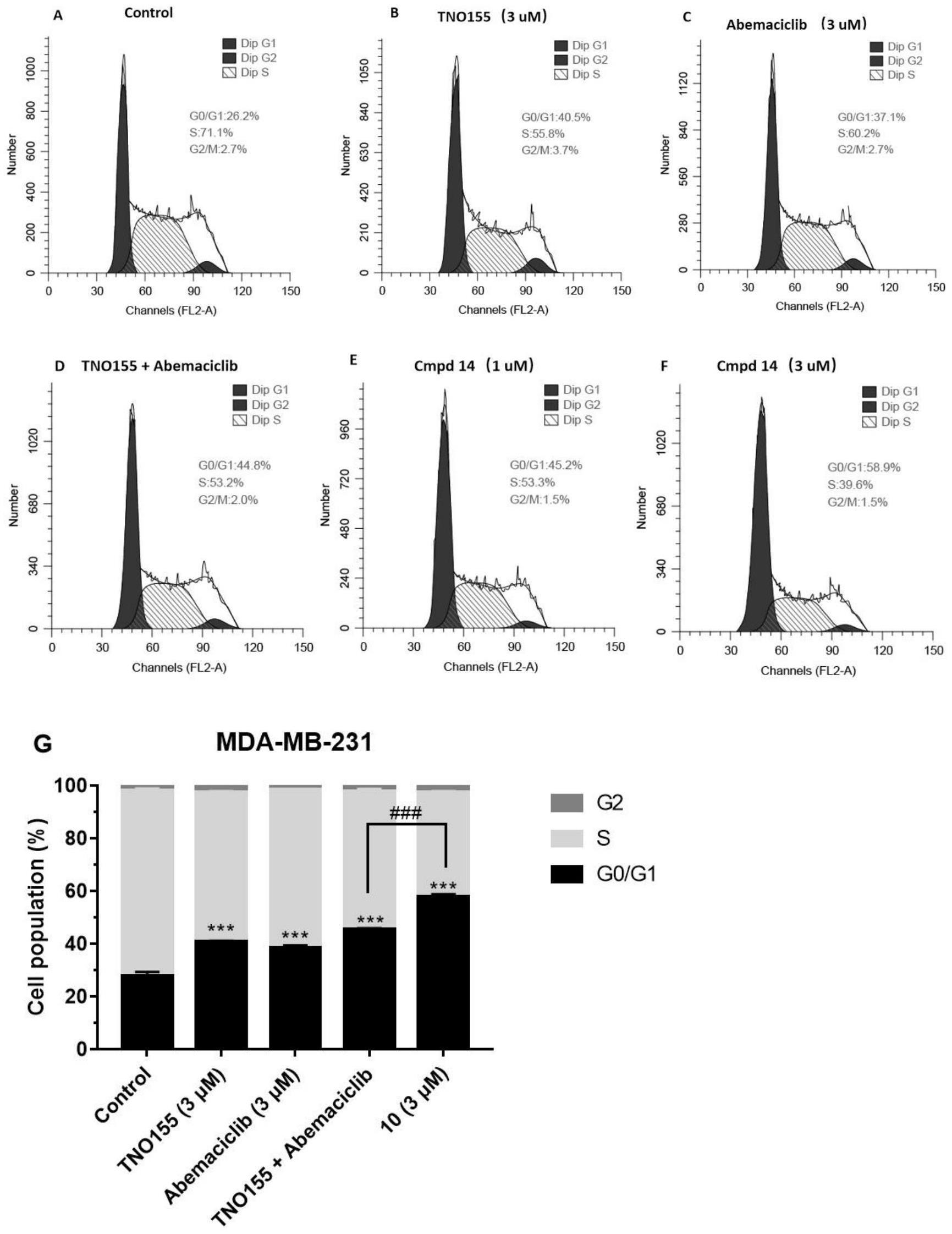
51、所述的芳基螺环类化合物、所述的药物组合物在制备治疗抗肿瘤药物、作为抗肿瘤药物的前药或作为抗肿瘤药物的中间体中应用。
52、设计思路:
53、
54、在双靶点药物设计中,药效团融合的设计方法往往可以避免高分子量及代谢差等问题。在shp2/cdk4双靶点抑制剂设计中,我们观察二者均具有吡啶环,且修饰均处于溶剂区边缘,于是创新性地将二者的药效团和连接基团合并。由于该融合型双靶点抑制剂较难平衡其在两个靶点之前的活性,我们简单探索不同药效团和连接基团对抑制活性的影响,得到化合物1-26。
55、sar分析发现,通过吡啶环对位连接两个药效团的化合物可以保持cdk4活性,但会降低shp2活性;通过吡啶环间位连接两个药效团的化合物可以保持shp2活性,但会降低cdk活性;abemaciclib的药效团整体效果要比ribociclib效果好;总之,我们通过合成26个化合物探索了shp2/cdk4双靶点抑制剂的sar,并获得多个具有双靶点抑制活性的化合物。
56、有益效果
57、1.本发明基于通过药效团融合和合理药物设计,制备出一类新型shp2与cdk4/6双靶点抑制剂,以解决目前shp2抑制剂和cdk4/6抑制剂耐药、药效差等问题。
58、2.在体外酶活和抗增殖活性测试中,化合物10、14、19、23、25、26显示出较好的双靶点抑制活性;在细胞周期实验中,化合物14显示出较好的周期g0/g1期阻滞作用。
59、3.具体实施例结构如下:
60、
61、
62、
- 还没有人留言评论。精彩留言会获得点赞!