一种可逆BTK抑制剂及其合成方法和应用
一种可逆btk抑制剂及其合成方法和应用
技术领域
1.本发明涉及小分子药物领域,具体地,本发明提供一种具有蛋白酪氨酸激酶(btk)抑制活性的新型分子,以及此类化合物的合成和应用。
背景技术:
2.据世界卫生组织统计,淋巴瘤发病率的年增长率为5%~7%,每年死亡人数超过20万,目前,我国淋巴瘤发病率的年增长率为3%~5%,每年的新发病例约10万,已经成为第八大高发性恶性肿瘤。
3.布鲁顿酪氨酸激酶(bruton’s tyrosine kinase,btk)属于非受体酪氨酸激酶tec家族,是一种膜结合蛋白,存在于除t细胞和自然杀伤细胞外的所有造血细胞中。btk是b细胞受体通路的重要信号分子,在b细胞的各个发育阶段表达,参与调控b细胞的增殖、分化与凋亡,在恶性b细胞的生存及扩散中起着重要作用。因此,btk是目前临床治疗b细胞类肿瘤及b细胞类免疫疾病的研究热点。
4.btk抑制剂可通过抑制bcr信号通路的过度激活从而抑制淋巴细胞增殖,对b细胞恶性肿瘤及自身免疫性疾病的治疗展现出极好的治疗前景。2013年,依鲁替尼被fda批准为第一个有效的btk选择性抑制剂,突破性治疗用于慢性淋巴细胞白血病、套细胞淋巴瘤、小淋巴细胞淋巴瘤、华氏巨球蛋白血症、边缘区淋巴瘤和移植物宿主病的治疗,具有划时代的意义,使btk成为一个有前途的治疗靶点。
5.虽然已有多个共价结合小分子btk抑制剂被应用于临床,如依鲁替尼、阿卡替尼和泽布替尼等,但随着接受治疗的患者不断增多和随访时间的延长,以及cys481残基的突变引起的对依鲁替尼的耐药性,引发人们对共价抑制剂的michael受体部分的思考和担忧。所以人们普遍认为,与不可逆抑制剂相比,可逆抑制剂更有可能在b细胞恶性肿瘤和自身免疫性疾病患者中具有较低的毒性风险。
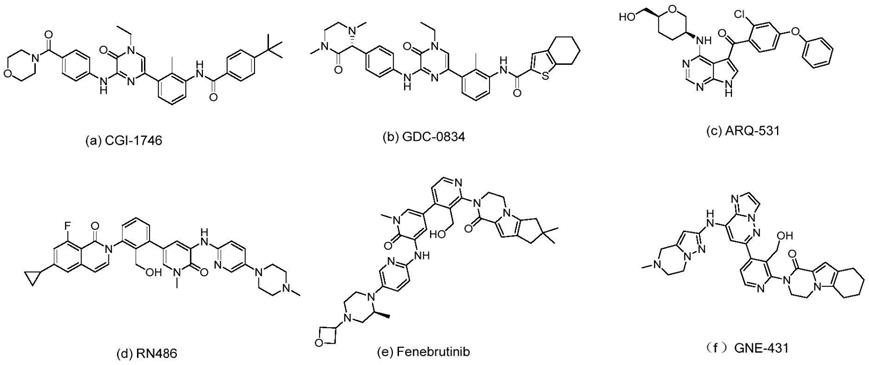
6.cgi-1746是第一个报道的与btk特异性非活性构象结合的化合物,具有极高的选择性的atp竞争性的可逆抑制剂。cgi-1746可同时抑制btk两个磷酸化途径,分子可以结合在btk非活性构象的结合口袋中,并且具有一定的稳定性。gdc-0834对btk以外的受体无活性,是一种高选择性、非共价、可逆的btk抑制剂,其体外ic
50
值为0.060μm。gdc-0834在大鼠胶原诱导的关节炎(cia)模型中显示出功效,导致踝关节肿胀的剂量依赖性减少和形态学病理学的减轻,被视为风湿性关节炎的潜在治疗药物。arq-531是一种抑制btk野生型和cys481突变型的候选药物,是一种口服、可逆的btk抑制剂。在临床前研究中,arq-531表现出较高的口服生物利用度和良好的药代动力学、代谢特性。rn-486是一种高选择性的btk抑制剂,可以抑制bcr介导的活化诱导因子cd69的释放。除此之外,部分可逆抑制剂还可以解决btk激酶中cys481位半胱氨酸突变造成的耐药性问题,例如arq531,gne-431等化合物均对突变的btk蛋白激酶有明显抑制作用。可逆btk抑制剂如下所示:
[0007][0008]
然而,与不可逆的共价抑制剂相比,可逆性btk抑制剂的发展进展缓慢,到目前为止,可逆抑制剂还处于临床试验阶段。考虑到btk治疗自身免疫性疾病的前景,迫切需要开发具有良好药效和药代动力学性质佳的新型可逆抑制剂。
技术实现要素:
[0009]
本发明的目的是针对现有技术的不足,提供一种具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物及其药学上可用的盐、其合成方法和应用。
[0010]
本发明的目的是通过以下技术方案来实现的:一种具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物,具有如下结构:
[0011][0012]
其中,r选自以下结构:
[0013][0014]
x独立地为氢、羟基、氨基或卤原子。
[0015]
进一步地,所述的具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物,选自以下结构:
[0016]
x独立地为氢或氟原子。
[0017]
更具休地,所述的一种具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物,选自以下结构:
[0018][0019][0020]
所有化合物均表现出对btk蛋白及btk突变株(btkc481s)的抑制活性,且对突变株的活性强于野生型。其中,化合物11活性最好,对btk蛋白的抑制活性处于nm级别;化合物6对野生型及突变型均有较好的活性。
[0021]
本发明所述的可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物可以单独使用,也可以用常规的方法制备成药学上可接受的盐使用,所述药学上可接受的盐
为盐酸盐、氢溴酸盐、氢碘酸盐、硫酸盐、硫酸氢盐、磷酸盐、乙酸盐、丙酸盐、丁酸盐、草酸盐、酒石酸盐、甲磺酸盐、对甲苯磺酸盐、富马酸盐、牛磺酸盐、柠檬酸盐、琥珀酸盐,或其混合盐。
[0022]
本发明同时提供一种具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物的制备方法,具体为:
[0023]
步骤a:以对溴苯甲酸或其取代物为起始原料,与嘧啶胺在缩合剂edc
·
hcl/hobt和n,n-二异丙基乙胺作用下通过缩合反应得到n-嘧啶基-4-溴苯甲酰胺;
[0024]
步骤b:上述得到的n-嘧啶基-4-溴苯甲酰胺与联硼酸频那醇酯通过宫浦偶联得到相应的硼酸酯产物;
[0025]
步骤c:原料4-氨基-3-碘-1h-唑咯并[3,4-d]嘧啶与原料(s)-1-叔丁氧羰基-3-羟基哌啶经mitsunobu反应得到(3r)-1-boc-3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶;
[0026]
步骤d:之后,(3r)-1-boc-3-(4-氨基-3-碘-1h-吡唑并[3,4-d]嘧啶-1-基)哌啶与前述得到的硼酸酯产物经suzuki反应得到偶联产物;
[0027]
步骤e:偶联产物在3m的hcl-ea溶液条件下脱去boc保护基得到哌啶类化合物;
[0028]
步骤f:哌啶类化合物经缩合或取代反应得到目标产物。
[0029]
反应方程式如下:
[0030][0031]
其中x、r与前面一致。
[0032]
本发明还提供了一种所述具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物及其在药学上可接受的盐在药学上的应用,该应用具体为:用于制备预防或治疗因btk蛋白表达过多所引起的疾病的药物制剂,所述的疾病为淋巴瘤,包括慢性淋巴细胞白血病、b细胞淋巴瘤、套细胞淋巴瘤、淋巴浆细胞淋巴瘤、弥漫性大b细胞淋巴瘤、非霍奇金淋巴瘤、滤泡中心淋巴瘤、边缘区b细胞淋巴瘤,或者为自体免疫疾病,包括类风湿关节炎、系统性红斑狼疮、强直性脊柱炎、银屑病等。
[0033]
与现有技术相比,本发明具有如下优点:
[0034]
本发明提供的可逆性btk抑制剂能够竞争性的占据atp与激酶的结合空腔而发挥
抑制作用,且不依赖于与cys481的相互作用,与atp铰链区的结合是通过较弱的、可逆性的作用力如氢键、范德华力和疏水作用力等来实现的,随着药物代谢的进行,药物与酶进行结合和脱离,以达到平衡。因而,大大减少了获得性耐药等副作用。故而,本发明具有可逆性btk抑制活性的恶唑并[4,5-b]吡啶结构的小分子化合物及其在药学上可接受的盐可能在制备预防或治疗因btk蛋白过度增长而引起的疾病的药物中有所应用,包括b细胞恶性肿瘤、哮喘和类风湿性关节炎等。
具体实施方式
[0035]
下面结合实施例对本发明的结构、制备方法及在制备预防或治疗因微管蛋白表达过多所引起的疾病的药物制剂方面的应用作进一步阐述,但不限制本发明。
[0036]
样品的分析数据由以下仪器测定:
[0037]
温度计未经校正;bruker drx400核磁共振仪;安捷伦5975型质谱仪;bruker vector 22红外光谱仪。
[0038]
实施例1.中间体1-1的合成
[0039][0040]
称取对溴苯甲酸1.31g(6.52mmol)置于10ml单颈瓶中,加入5ml二氯甲烷,冰浴条件下依次加入edc
·
hcl 1.25g(6.52mmol),dipea2.81ml(16.14mmol)搅拌5min,保持0℃加入2-氨基吡啶0.50g(5.38mmol)后置于室温下反应过夜。经tlc监测反应完全,减压除去二氯甲烷,加饱和食盐水(200ml
×
3)和乙酸乙酯(150ml)萃取,有机相合并,经无水硫酸钠干燥,旋干后干法拌样过柱得到得到棕色粉末状固体,收率约为89.1%。ms(esi):m/z=276.99[m+h]
+
;1h nmr(400mhz,cdcl3)δ8.93(s,1h),8.63(d,j=1.3hz,1h),8.46(d,j=2.5hz,1h),8.35(dd,j=2.5,1.5hz,1h),7.60
–
7.58(m,2h),7.55
–
7.53(m,2h).
[0041]
实施例2.中间体1-2的合成
[0042][0043]
依次称取实施例1产物1.00g(3.61mmol),联硼酸频那醇酯1.10g(4.33mmol),[1,1'-双(二苯基膦)二茂铁]二氯化钯二氯甲烷络合物79mg(0.11mmol),醋酸钾1.06g(10.83mmol)置于25ml三颈瓶中,n2保护,之后注射器加入四氢呋喃溶液,回流过夜。点板原料反应完毕后,反应液冷却至室温后用硅藻土抽滤,并用四氢呋喃冲洗2~3次,滤液浓缩后加水加乙酸乙酯萃取,有机层用水洗2~3次,减压回收后得到粗品,经柱层析分离纯化后得
到淡棕色粉末状固体,收率约为92.6%1h nmr(400mhz,dmso-d6)δ10.89(s,1h),8.40(d,j=4.8hz,1h),8.19(d,j=8.4hz,1h),8.02(d,j=8.2hz,2h),7.88
–
7.82(m,1h),7.79(d,j=8.2hz,2h),7.24
–
7.15(m,1h),1.32(s,12h).
[0044]
实施例3.中间体2-1的合成
[0045][0046]
称取3-氟4-溴苯甲酸1.00g(4.57mol)置于单颈烧瓶中,加入5ml二氯甲烷,反应瓶置于冰浴条件下后依次加入hatu 1.74g(4.57mmol),dipea 2.39ml(13.71mmol)和2-氨基吡啶1.13g(3.81mmol),然后转移至室温反应过夜。通过tlc确定反应毕,减压除去溶剂二氯甲烷,加饱和食盐水(200ml
×
3)和乙酸乙酯250ml萃取,有机相合并,经无水硫酸钠干燥,浓缩后干法拌样过柱,pe润柱,ea:pe=1:4洗脱得到1.12g淡棕色粉末状固体,收率约为74.6%。ms(esi):m/z=294.98[m+h]
+
;1h nmr(400mhz,dmso-d6)δ10.99(s,1h),8.41(ddd,j=4.8,1.9,0.8hz,1h),8.17(d,j=8.4hz,1h),8.02(dd,j=9.8,1.9hz,1h),7.93
–
7.79(m,3h),7.25
–
7.14(m,1h).
[0047]
实施例4.中间体2-2的合成
[0048][0049][0050]
操作同实施例2,中间体2-1代替中间体1-1,得到白色固体,收率约为91.6%;1h nmr(400mhz,dmso-d6)δ10.93(d,j=32.2hz,1h),8.47
–
8.35(m,2h),8.18(d,j=8.4hz,1h),7.90
–
7.69(m,3h),7.24
–
7.14(m,1h),1.33(s,12h).
[0051]
实施例5.中间体2-3的合成
[0052][0053]
分别称取4-氨基-3-碘-1h-吡唑[3,4-d]嘧啶500mg(1.92mmol),(r)-1-叔丁氧羰基-3-羟基哌啶774mg(3.83mmol),三苯基膦755mg(2.88mmol)加入到三颈瓶中,n2保护,冰浴条件下用注射器加入无水thf,再慢慢滴加diad的无水thf溶液,后转移至室温反应过夜。tlc点板原料反应完后,减压除去thf,干法拌样过柱,pe润柱可冲出黄色的diad,之后梯度洗脱得到708mg白色粉末状固体,即中间体2-3,收率约为83.1%。ms(esi):m/z=467.24[m+
na]
+
;1h nmr(400mhz,dmso-d6)δ8.21(s,1h),4.65
–
4.53(m,1h),3.88(t,j=61.6hz,2h),2.98(s,1h),2.19
–
2.08(m,1h),2.07
–
1.98(m,1h),1.87(s,1h),1.54(dt,j=21.8,7.5hz,2h),1.32(s,9h).
[0054]
实施例6.中间体1-3的合成
[0055][0056]
依次称取实施例1 436mg(1.35mmol),实施例5 500mg(1.13mmol),pd(dppf)cl2 25mg(0.03mmol),k2co
3 468mg(3.39mmol)加入到10ml三颈瓶中,氮气保护,冰浴条件下加入1,4二氧六环:水(3:1)的混合溶液,然后转移至105℃开始回流反应。8h后tlc监测反应进展,反应毕反应液先冷却至室温,用硅藻土抽滤除去pd(dppf)cl2,并用ea冲洗2~3次,然后滤液加水(100ml
×
4)和100ml乙酸乙酯萃取,有机相用无水na2so4干燥后经梯度洗脱得到淡棕色粉末状固体1-3,收率约为73.4%;ms(esi):m/z=515.25[m+h]
+
;1h nmr(400mhz,cdcl3)δ9.10(s,1h),8.44(d,j=8.4hz,1h),8.37(s,1h),8.32(d,j=4.9hz,1h),8.14(d,j=8.3hz,2h),7.87(d,j=8.2hz,2h),7.85
–
7.77(m,1h),7.13(dd,j=7.3,5.0hz,1h),4.87
–
4.73(m,1h),3.63(dd,j=13.5,6.4hz,2h),2.90(t,j=11.5hz,2h),2.26
–
2.17(m,2h),1.84
–
1.64(m,2h),1.46(s,12h).
[0057]
实施例7.中间体1-4的合成
[0058][0059]
称取实施例6 300mg(0.58mmol)于单颈瓶中,加入2ml乙酸乙酯,冰浴条件下滴加3ml 3mol hcl的乙酸乙酯溶液,滴加过程中反应液变浑浊,滴加完毕后反应液转移至室温反应2h。tlc监测反应毕后,反应液滤纸过滤,滤饼即为产物盐酸盐。后将滤饼置于100ml 0℃的k2co3水溶液中,用乙酸乙酯(120ml
×
4)萃取,有机相合并,无水硫酸钠干燥,得到淡棕
色粉末状固体1-4,收率约为61.9%;ms(esi):m/z=437.20[m+h]
+
;1h nmr(400mhz,dmso-d6)δ11.66(s,1h),9.75(d,j=10.1hz,1h),9.51(d,j=10.9hz,1h),8.64(s,1h),8.49(d,j=4.8hz,1h),8.36
–
8.26(m,2h),8.10(t,j=7.7hz,1h),7.86(d,j=8.1hz,1h),7.47
–
7.30(m,1h),5.26
–
5.10(m,1h),3.54(d,j=10.7hz,1h),3.50
–
3.38(m,2h),3.31(d,j=11.5hz,1h),3.12
–
2.93(m,1h),2.15(d,j=5.2hz,2h),1.05(t,j=7.0hz,1h).
[0060]
实施例8.化合物
ⅰ‑
1的合成
[0061][0062]
将醋酸8mg(0.09mmol)溶于二氯甲烷,依次加入edc
·
hcl 14mg(0.08mmol),三乙胺21ul,搅拌5分钟后加入中间体1-4 30mg(0.08mmol),室温反应。反应3h后,tlc监测反应毕,反应液浓缩除去二氯甲烷,后加50ml ea溶解,加水70ml萃取,后有机层用1n的稀hcl溶液(70ml
×
2)萃取,饱和nacl溶液70ml萃取一次,ea层用无水na2so4干燥后旋出溶剂得粗品,经梯度洗脱后得到白色固体
ⅰ‑
1,收率约为71.6%;ms(esi):m/z=457.20[m+h]
+
;1h nmr(400mhz,acetone-d6)δ8.39(t,j=7.5hz,2h),8.32
–
8.22(m,3h),7.91(dt,j=15.8,7.5hz,3h),7.21
–
7.15(m,1h),3.80
–
3.69(m,1h),3.57(q,j=7.0hz,1h),3.17(q,j=7.2hz,2h),2.18
–
2.10(m,3h),1.95(s,3h),1.29(t,j=7.2hz,1h).
13
c nmr(101mhz,acetone-d6)δ182.54,168.95,158.34,148.23,147.91,138.56,138.23,136.68,134.17,132.46,128.74,128.40,114.39,114.02,97.96,46.05,20.50.
[0063]
实施例9.化合物
ⅰ‑
2的合成:
[0064][0065][0066]
操作同实施例8,三氟乙酸代替乙酸,得到淡棕色半固体
ⅰ‑
2,收率约为52.4%;ms(esi):m/z=511.18[m+h]
+
;1h nmr(400mhz,cdcl3)δ8.96(s,1h),8.47
–
8.37(m,2h),8.33
(d,j=2.5hz,1h),8.12(dd,j=8.2,5.3hz,2h),7.85(dd,j=8.3,4.8hz,2h),7.12(td,j=6.5,1.4hz,1h),4.94
–
4.79(m,1h),4.05(dd,j=13.0,4.1hz,1h),3.92
–
3.81(m,1h),3.73(dd,j=13.1,10.6hz,1h),3.36
–
3.15(m,2h),2.86
–
2.75(m,1h),2.40(dd,j=12.0,3.5hz,1h),1.82
–
1.62(m,1h).
13
c nmr(101mhz,cdcl3)δ182.54,168.95,158.34,148.23,147.91,138.56,138.23,136.68,134.17,132.46,128.74,128.40,114.39,114.02,97.96,46.05,20.50.
[0067]
实施例10.化合物
ⅰ‑
3的合成:
[0068][0069]
称取中间体4-9的盐酸盐50mg(0.12mmol)置于封管中,加三氟乙醇溶解,冰浴条件下加入环氧丙烷10μl(0.12mmol),然后置于80℃反应3h,tlc点板反应毕,减压除去三氟乙醇和多余的环氧丙烷,得到棕色油状液体
ⅰ‑
3,收率约为90.3%;ms(esi):m/z=473.25[m+h]
+
;1h nmr(400mhz,dmso-d6)δ10.92(s,1h),8.42(dd,j=4.8,1.1hz,1h),8.29(s,1h),8.22(t,j=7.9hz,3h),7.92
–
7.84(m,1h),7.80(d,j=8.3hz,2h),7.24
–
7.15(m,1h),3.90
–
3.82(m,1h),3.40(d,j=10.1hz,1h),3.29(d,j=7.0hz,3h),2.65
–
2.46(m,3h),2.15
–
2.04(m,2h),1.90(dd,j=9.3,5.7hz,2h),1.60(dd,j=24.5,13.2hz,3h).
13
c nmr(101mhz,dmso-d6)δ170.67,165.95,159.13,155.51,153.63,143.10,139.34,136.48,134.74,123.72,120.22,113.72,97.34,67.51,65.47,63.20,59.71,56.19,30.26,19.96.
[0070]
实施例11.化合物
ⅰ‑
4的合成:
[0071][0072]
操作同实施例10,缩水甘油代替环氧丙烷,得到棕色油状液体
ⅰ‑
4,收率约为91.4%;ms(esi):m/z=489.23[m+h]
+
;1h nmr(400mhz,dmso-d6)δ8.42(s,1h),8.35(dd,j=6.8,4.3hz,1h),8.23(d,j=8.3hz,2h),7.87(dd,j=10.8,4.6hz,1h),7.81(t,j=7.3hz,
2h),7.25
–
7.14(m,2h),3.85
–
3.72(m,1h),3.43(d,j=4.0hz,2h),3.09(t,j=5.2hz,2h),2.65
–
2.56(m,1h),2.17
–
2.09(m,2h),2.07
–
1.96(m,2h),1.87(dd,j=9.1,5.9hz,2h),1.53(dd,j=24.3,12.8hz,2h).
13
c nmr(101mhz,dmso-d6)δ170.95,158.71,148.46,138.69,129.29,128.68,115.25,98.05,72.97,69.69,64.97,62.70,47.63,46.86,27.38,21.23.
[0073]
实施例12.化合物
ⅰ‑
5的合成:
[0074][0075]
操作同实施例8,boc-l-丙氨酸代替乙酸,得到淡棕色固体
ⅰ‑
5,收率约为72.6%;ms(esi):m/z=586.28[m+h]
+
;1h nmr(400mhz,dmso-d6)δ10.91(s,1h),8.42(s,1h),8.29(d,j=8.3hz,1h),8.22(t,j=8.2hz,2h),7.92
–
7.85(m,1h),7.80(d,j=8.3hz,2h),7.25
–
7.13(m,2h),4.54
–
4.44(m,1h),4.03(q,j=7.1hz,1h),3.68
–
3.56(m,2h),3.14(t,j=6.6hz,1h),2.17(dd,j=13.5,10.3hz,1h),1.99(dd,j=10.6,4.2hz,2h),1.91(d,j=6.4hz,3h),1.67
–
1.52(m,2h),1.36(s,9h).
[0076]
实施例13.化合物
ⅰ‑
6的合成:
[0077][0078]
操作同实施例8,l-丙氨酸代替醋酸,得到淡棕色半固体
ⅰ‑
6,收率约为81.4%;ms(esi):m/z=486.23[m+h]
+
;1h nmr(400mhz,dmso-d6)δ10.92(s,1h),8.78(s,1h),8.44(d,j=3.9hz,1h),8.31(d,j=8.5hz,1h),8.25(t,j=7.9hz,2h),7.68(d,j=8.3hz,2h),7.23
–
7.15(m,2h),4.57
–
4.42(m,1h),3.74(q,j=5.8hz,1h),3.69(d,j=7.0hz,1h),3.54(d,j=10.2hz,1h),3.13(t,j=14.4,2h),2.17
–
1.86(m,2h),1.68
–
1.58(m,2h),1.27(td,j=6.9,10.5hz,3h).
13
c nmr(101mhz,dmso-d6)δ173.69,164.58,158.46,154.32,152.74,148.65,142.51,139.98,137.26,134.70,131.47,128.93,117.46,115.82,101.42,64.58,51.46,47.35,43.76,29.73,20.97,18.36.
[0079]
实施例14.化合物
ⅰ‑
7的合成:
[0080][0081]
操作同实施例8,中间体7-9代替中间体1-6,醋酸代替2-甲基丙烯酸1-7,得到白色半固体
ⅰ‑
7,收率约为67.2%;ms(esi):m/z=475.19[m+h]
+
;1h nmr(400mhz,acetone-d6)δ9.96(s,1h),8.37(d,j=7.8hz,2h),8.26(s,1h),8.11(d,j=8.0hz,1h),8.04(d,j=10.8hz,1h),7.84(dt,j=21.5,7.8hz,2h),7.21
–
7.14(m,1h),3.99
–
3.88(m,1h),3.63(t,j=6.1hz,1h),3.40(dd,j=14.7,7.3hz,1h),3.21(dd,j=26.8,13.1hz,2h),2.11(d,j=8.7hz,2h),2.06(s,3h),1.79(t,j=6.1hz,2h).
13
c nmr(101mhz,acetone-d6)δ168.57,168.36,164.14,161.02,158.55,158.40,156.17,156.10,154.24,152.19,148.32,148.00,138.02,137.37,132.16,124.82,120.11,119.93,116.24,114.64,114.19,99.52,54.94,53.45,52.77,20.79,17.56.
[0082]
实施例15.化合物
ⅰ‑
8的合成:
[0083][0084]
操作同实施例8,中间体2-2代替中间体1-2,三氟乙酸代替醋酸,得到白色半固体
ⅰ‑
8,收率约为53.9%;ms(esi):m/z=529.16[m+h]
+
;1h nmr(400mhz,cdcl3)δ8.95(s,1h),8.39(t,j=10.2hz,2h),8.32(dd,j=4.0,3.2hz,2h),8.10(dd,j=9.4,4.1hz,3h),3.93
–
3.88(m,1h),3.66(t,j=6.1hz,2h),3.45(dd,j=13.5,7.9hz,2h),3.29(dd,j=16.3,12.5hz,2h),2.12(t,j=7.5hz,1h),2.03(t,j=5.5hz,1h).
13
c nmr(101mhz,cdcl3)δ163.65,158.23,155.64,153.54,151.62,145.64,141.46,129.10,126.14,124.07,118.38,114.29,112.86,111.04,99.49,65.08,52.68,51.29,30.65,24.39.
[0085]
实施例16.化合物
ⅰ‑
9的合成:
[0086][0087]
操作同实施例10,中间体2-2代替中间体1-2,得到棕色油状液体
ⅰ‑
9,收率约为63.8%;ms(esi):m/z=491.22[m+h]
+
;1h nmr(400mhz,dmso-d6)δ11.03(s,1h),8.44(d,j=3.6hz,1h),8.35
–
8.17(m,2h),8.14
–
8.00(m,2h),7.90(t,j=7.2hz,1h),7.72(t,j=7.6hz,1h),7.29
–
7.16(m,1h),4.03
–
3.90(m,1h),3.40(d,j=10.0hz,2h),3.11(dd,j=17.6,8.7hz,1h),2.71
–
2.60(m,2h),2.53
–
2.41(m,2h),2.04(d,j=10.1hz,1h),1.88(d,j=16.9hz,2h),1.65
–
1.52(m,1h),1.08(d,j=5.9hz,3h).
13
c nmr(101mhz,dmso-d6)δ172.59,164.81,160.87,158.48,158.41,156.35,154.09,154.07,152.38,148.50,138.74,137.82,136.91,132.04,124.93,124.26,120.63,116.72,116.49,115.39,99.27,29.18,22.09.
[0088]
实施例17.化合物
ⅰ‑
10的合成:
[0089][0090]
操作同实施例10,中间体2-2代替中间体1-2,缩水甘油代替环氧丙烷,得到棕色油状液体
ⅰ‑
10,收率约为63.8%;ms(esi):m/z=507.23[m+h]
+
;1h nmr(400mhz,dmso-d6)δ8.44(s,1h),8.23(d,j=8.3hz,1h),8.06(dd,j=11.9,5.7hz,2h),7.94
–
7.81(m,2h),7.73(dt,j=12.8,7.8hz,1h),7.26
–
7.18(m,1h),4.06
–
3.89(m,1h),3.57(dd,j=4.0,2.8hz,4h),2.63(m,1h),2.29
–
2.15(m,2h),2.15
–
2.04(m,2h),1.90(dt,j=9.3,5.7hz,2h),1.60(dt,j=24.5,13.2hz,2h).
13
c nmr(101mhz,dmso-d6)δ170.94,158.44,156.21,154.89,152.40,148.46,138.74,127.20,124.42,115.38,115.34,108.12,99.24,72.63,69.57,64.48,59.47,53.84,21.21.
[0091]
实施例18.化合物
ⅰ‑
11的合成:
[0092][0093]
操作同实施例8,中间体2-2代替中间体1-2,马来酰胺酸代替醋酸,得到白色固体
ⅰ‑
11,收率约为63.8%;ms(esi):m/z=512.20[m+h]
+
;1h nmr(400mhz,acetone-d6)δ9.91(s,1h),8.37(d,j=7.4hz,2h),8.27(s,1h),8.12(d,j=7.2hz,1h),8.04(s,1h),7.93
–
7.73(m,2h),7.37(dd,j=30.8,11.7hz,1h),6.58(d,j=13.1hz,1h),6.04(d,j=11.7hz,1h),3.94(t,j=7.8hz,1h),3.78
–
3.63(m,1h),3.54(t,j=7.0hz,1h),2.48
–
2.34(m,2h),2.28(dd,j=10.7,6.2hz,1h),1.92
–
1.65(m,2h),1.12(d,j=6.9hz,1h).
13
c nmr(101mhz,acetone-d6)δ164.11,162.26,158.37,156.21,155.83,152.13,148.34,141.97,138.00,137.54,124.65,123.94,120.12,119.92,116.00,115.69,114.46,106.30,99.45,59.68,56.84,29.19,28.42,17.84.
[0094]
实施例19.体外酶抑制实验
[0095]
1.化合物配制
[0096]
称取以上化合物1mg溶解在100%dmso-d6中,配制成10mm储存液,氮气柜中避光保存。
[0097]
2.激酶反应过程
[0098]
(1)配制1
×
kinase buffer。
[0099]
(2)化合物浓度梯度的配制:受试化合物起始测试浓度为2400nm,7个浓度,复孔
[0100]
检测。在384source板中稀释成100倍终浓度的100%dmso-d6溶液。使用分液器echo 550向目的板3575板转移250nl 100倍终浓度的化合物。
[0101]
(3)用1
×
kinase buffer配制2.5倍终浓度的激酶溶液。
[0102]
(4)在化合物孔和阳性对照孔分别加10μl的2.5倍终浓度的激酶溶液;在阴性对照
[0103]
孔中加10μl的1
×
kinase buffer。
[0104]
(5)1000rpm离心30秒,反应板振荡混匀后室温孵育10分钟。
[0105]
(6)用1
×
kinase buffer配制5/3倍终浓度的atp和kinase substrate 2的混合溶液。
[0106]
(7)加入15μl的5/3倍终浓度的atp和底物的混合溶液,起始反应。
[0107]
(8)将384孔板1000rpm离心30秒,振荡混匀后室温孵育30分钟。
[0108]
(9)加入30μl终止检测液停止激酶反应,1000rpm离心30秒,振荡混匀。
[0109]
(10)用caliper ez reader读取转化率。
[0110]
结果见表1。
[0111]
表1.化合物
ⅰ‑
1~
ⅰ‑
10的btk抑制活性结果
[0112][0113]
[0114][0115]ano detection
[0116]
由表1可见,所有化合物均表现出对btk蛋白及btk突变株(btkc481s)的抑制活性,且对突变株的活性强于野生型。相对而言,化合物
ⅰ‑
11活性最好,对btk蛋白的抑制活性处于nm级别;化合物
ⅰ‑
6(ic
50
=42.0nm,ic
50c481s
=31.0nm)对野生型及突变型均有较好的活性。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1