两面针中两对二氢苯并菲啶生物碱类化合物及其提取方法和应用
1.本发明涉及药物提取分离技术领域,具体涉及两面针中两对二氢苯并菲啶生物碱类化合物及其提取方法和应用。
背景技术:
2.中药两面针来源于芸香科植物两面针[zanthoxylum nitidum(roxb.)dc.]的根或枝叶。分布于中南地区及福建,台湾,云南,四川等地,全年可采收。两面针为历代民间用药,中医传统认为为其具有活血,行气,祛风,止痛和消肿的功能。主要用于风湿疼痛,风寒湿痺引起的胃痛,腹痛,牙痛,跌打损伤以及毒蛇咬伤等症用。近年来,因发现两面针具有良好的镇痛和抗癌作用,渐被广泛应用。
[0003]
现代药理学研究表明两面针植物含有多种类型生物碱,主要为苯并菲啶类、喹啉类和异喹啉类生物碱。具有多种特殊而显著的生理作用,主要有抑制血小板凝集、细胞毒活性、抑制dna异构酶和选择性抑菌作用。
[0004]
目前对两面针化学成分的研究主要集中在生物碱类化合物,但目前还未见有本发明所分离的这两对6-一取代二氢苯并菲啶生物碱类化合物的相关报道。
技术实现要素:
[0005]
本发明的发明目的在于:针对上述存在的问题,提供两面针中两对二氢苯并菲啶生物碱类化合物及其提取方法和应用,本发明通过化学分离得到两对二氢苯并菲啶生物碱类化合物,该两对二氢苯并菲啶生物碱类化合物未曾被公开报道,具有体外抗肿瘤活性,具有较好的潜在药用价值,有望用于各种抗肿瘤药物的制备。
[0006]
为了实现上述目的,本发明采用的技术方案如下:
[0007]
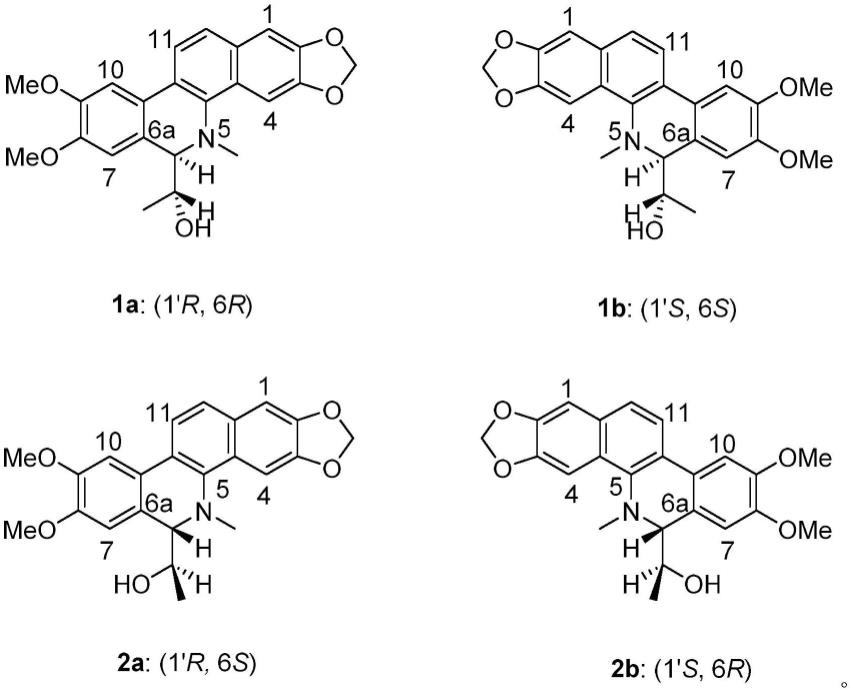
本发明首先提供两面针中两对二氢苯并菲啶生物碱类化合物,所述化合物为下式1a、1b、2a、2b所示化合物或其药学上可接受的盐:
[0008][0009]
本发明还提供上述两面针中两对二氢苯并菲啶生物碱类化合物的提取方法,包括以下步骤:
[0010]
(1)以两面针为原料,提取获得两面针提取物;
[0011]
(2)将两面针提取物溶于质量分数为3%的酒石酸溶液,滤去不溶物a后将滤液用乙酸乙酯萃取,分离得乙酸乙酯萃取物和酸水不溶物;
[0012]
(3)将乙酸乙酯萃取物和酸水不溶物合并,依次用石油醚、乙酸乙酯、正丁醇萃取,浓缩得石油醚部位、乙酸乙酯部位、正丁醇部位;
[0013]
(4)将纯化后的乙酸乙酯部位上硅胶柱层析,以二氯甲烷和甲醇组成的洗脱剂作为溶剂体系进行梯度洗脱,对馏分进行分析检测,收集含式1a、1b、2a、2b所示化合物的目标馏分,所得含目标化合物的目标馏分依次用反相色谱柱、葡聚糖凝胶柱、制备液相进行分离,即得到目标化合物;
[0014]
(5)通过手性柱对目标化合物进行拆分,即得到所述两对二氢苯并菲啶生物碱类化合物。
[0015]
上述提取方法中,步骤(1)中所述的两面针为花椒属芸香科植物zanthoxylum nitidum,优选根部为原料。
[0016]
上述提取方法中,获得岩黄连提取物的方法与现有技术相同,具体可以是以两面针为原料,提取所用的溶媒为体积浓度为70-100%甲醇或体积浓度为75-95%乙醇或者丙酮。提取的方式具体可以是现有常规的常温浸提、加热提取、超声波提取或回流提取等。
[0017]
上述提取方法中,步骤(2)中,当浸膏溶于3%的酒石酸溶液后,得浸膏溶液的ph=2-3;滤去不溶物a后滤液用乙酸乙酯萃取的次数为3-4次。
[0018]
上述提取方法中,步骤(3)中依次用石油醚、乙酸乙酯、正丁醇萃取,每种萃取液萃取的次数为3-4次。
[0019]
上述提取方法中,步骤(4)中,硅胶柱层析时,按照二氯甲烷和甲醇按体积比为100:1,80:1,50:1,40:1,30:1,20:1,15:1,10:1,8:1,5:1,2:1,1:1进行梯度洗脱,每个梯度浓度的洗脱剂体积为柱体积的3-4倍,每500ml馏分进行收集,对馏分进行分析检测。
[0020]
上述提取方法中,步骤(4)中,在用反相色谱柱分离时,洗脱剂由甲醇和水按不同的体积比组成的,按甲醇含量为20%,30%,40%,50%,60%,70%,80%,90%的梯度洗脱,每300ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0021]
在用葡聚糖凝胶柱分离时,洗脱剂为甲醇,每30ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0022]
在用制备液相分离时,流速为7ml/min,检测波长为210nm,分离条件为:流动相为:甲醇和水,在为甲醇:水的体积比为65:35时,收集保留时间为49.0min和51min的组分,分别为化合物1和化合物2。
[0023]
上述提取方法中,步骤(5)进行所述手性拆分时,流动相流速为0.7ml/min,色谱柱温度为30℃,检测波长为210nm;化合物1手性拆分的流动相为体积比是62:38的正己烷和异丙醇,采用等度洗脱方式,在保留时间为20.9min得到式1a化合物,在保留时间为33.8min得到式1b化合物;化合物2手性拆分的流动相为体积比是88:12的正己烷和异丙醇,采用等度洗脱方式,在保留时间为36.0min得到式2a化合物,在保留时间为38.7min得到式2b化合物。
[0024]
上述提取方法中,步骤(4)对馏分进行分析检测时,检测手段可以是tlc,优选为hplc,更优选为hplc-ms。
[0025]
本发明还保护上述化合物或其药学上可接受的盐在制备抗肿瘤药物中的应用。
[0026]
本发明还保护上述化合物或其药学上可接受的盐为活性成分制备的抗肿瘤药物。
[0027]
综上所述,由于采用了上述技术方案,本发明的有益效果是:
[0028]
本发明提供了两对二氢苯并菲啶生物碱类化合物,以及它们的制备方法和应用,这化合物的制备方法简单易操作。通过考察化合物对多种肿瘤的抑制作用,结果表明该化合物具有体外抗肿瘤活性,具有较好的潜在药用价值,有望用于各种抗肿瘤药物的制备。
具体实施方式
[0029]
为了更清楚地表达本发明,以下通过具体实施例对本发明作进一步说明。
[0030]
实施例1
[0031]
两对结构新颖的二氢苯并菲啶生物碱类化合物的制备方法,包括以下步骤:
[0032]
(1)取两面针干燥全株部分28kg,粉碎,用体积浓度为95%的乙醇为溶媒,回流提取3次,合并提取液,减压回收溶剂,得到两面针提取物浸膏3.4kg;
[0033]
(2)将两面针提取物浸膏溶于质量分数为3%的酒石酸溶液,调节浸膏溶液的ph=2;滤去不溶物a后将滤液用乙酸乙酯萃取3次,减压浓缩回收溶剂,得乙酸乙酯萃取物和酸水不溶物;
[0034]
(3)将乙酸乙酯萃取物和酸水不溶物合并,依次用石油醚、乙酸乙酯、正丁醇萃取,每种萃取液萃取的次数为3-4次,浓缩纯化得石油醚部位、乙酸乙酯部位、正丁醇部位;
[0035]
(4)将纯化后的乙酸乙酯部位上硅胶柱层析,按照二氯甲烷和甲醇按体积比为
100:1,80:1,50:1,40:1,30:1,20:1,15:1,10:1,8:1,5:1,2:1,1:1进行梯度洗脱,每个梯度浓度的洗脱剂体积为柱体积的3倍,每500ml馏分进行收集,分段收集样品,以tlc进行划段合瓶得到11段馏分,分段收集样品,以tlc进行划段合瓶得到11段馏分,用hplc-uv和hplc-ms分析检测,收集含式1a、1b、2a、2b所示化合物的目标馏分。
[0036]
所得含目标化合物的目标馏分用反相色谱柱进行分离,反相色谱柱分离的洗脱剂由甲醇和水按不同的体积比组成的,按甲醇含量为20%,30%,40%,50%,60%,70%,80%,90%的梯度洗脱,每300ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0037]
将目标馏份再用葡聚糖凝胶柱进行分离,葡聚糖凝胶柱分离的洗脱剂为甲醇,每30ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0038]
将目标馏份再用制备液相进行分离,制备液相分离的条件为:流动相为甲醇和水,流速为7ml/min,检测波长为210nm,分离条件是在甲醇:水的体积比为65:35时,收集保留时间为49.0min和51min的组分,分别为化合物1和化合物2。
[0039]
(5)通过手性柱对目标化合物进行拆分,进行所述手性拆分时,流动相流速为0.7ml/min,色谱柱温度为30℃,检测波长为210nm;化合物1手性拆分的流动相为体积比是62:38的正己烷和异丙醇,采用等度洗脱方式,在保留时间为20.9min得到式1a化合物,在保留时间为33.8min得到式1b化合物;化合物2手性拆分的流动相为体积比是88:12的正己烷和异丙醇,采用等度洗脱方式,在保留时间为36.0min得到式2a化合物,在保留时间为38.7min得到式2b化合物。
[0040]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b。
[0041]
实施例2
[0042]
两对结构新颖的二氢苯并菲啶生物碱类化合物的制备方法,包括以下步骤:
[0043]
(1)取两面针干燥全株部分27kg,粉碎,用体积浓度为75%的乙醇常温浸提3次,每次3h,合并提取液,减压回收溶剂,得到两面针提取物浸膏;
[0044]
(2)将两面针提取物浸膏溶于质量分数为3%的酒石酸溶液,调节浸膏溶液的ph=3;滤去不溶物a后将滤液用乙酸乙酯萃取3次,减压浓缩回收溶剂,得乙酸乙酯萃取物和酸水不溶物;
[0045]
(3)将乙酸乙酯萃取物和酸水不溶物合并,依次用石油醚、乙酸乙酯、正丁醇萃取,每种萃取液萃取的次数为3-4次,浓缩纯化得石油醚部位、乙酸乙酯部位、正丁醇部位;
[0046]
(4)将纯化后的乙酸乙酯部位上硅胶柱层析,按照二氯甲烷和甲醇按体积比为100:1,80:1,50:1,40:1,30:1,20:1,15:1,10:1,8:1,5:1,2:1,1:1进行梯度洗脱,每个梯度浓度的洗脱剂体积为柱体积的3-4倍,每500ml馏分进行收集,分段收集样品,以tlc进行划段合瓶得到11段馏分,用hplc-uv和hplc-ms分析检测,收集含式1a、1b、2a、2b所示化合物的目标馏分。
[0047]
所得含目标化合物的目标馏分用反相色谱柱进行分离,反相色谱柱分离的洗脱剂由甲醇和水按不同的体积比组成的,按甲醇含量为20%,30%,40%,50%,60%,70%,80%,90%的梯度洗脱,每300ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0048]
将目标馏份再用葡聚糖凝胶柱进行分离,葡聚糖凝胶柱分离的洗脱剂为甲醇,每30ml馏分进行收集,对馏分进行检测,收集目标馏份;
[0049]
将目标馏份再用制备液相进行分离,制备液相分离的条件为:流动相为甲醇和水,
流速为7ml/min,检测波长为210nm,分离条件是在甲醇:水的体积比为65:35时,收集保留时间为49.0min和51min的组分,分别为化合物1和化合物2。
[0050]
(5)通过手性柱对目标化合物进行拆分,进行所述手性拆分时,流动相流速为0.7ml/min,色谱柱温度为30℃,检测波长为210nm;化合物1手性拆分的流动相为体积比是62:38的正己烷和异丙醇,采用等度洗脱方式,在保留时间为20.9min得到式1a化合物,在保留时间为33.8min得到式1b化合物;化合物2手性拆分的流动相为体积比是88:12的正己烷和异丙醇,采用等度洗脱方式,在保留时间为36.0min得到式2a化合物,在保留时间为38.7min得到式2b化合物。
[0051]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b。
[0052]
实施例3
[0053]
重复实施例1,不同的是:将步骤(1)中的提取溶媒由体积浓度为95%的乙醇更改为100%的甲醇。
[0054]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b
[0055]
实施例4
[0056]
重复实施例2,不同的是:将步骤(1)中的提取溶媒由体积浓度为75%的乙醇更改为70%的甲醇,提取方式采用超声波提取。
[0057]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b。
[0058]
实施例5
[0059]
重复实施例1,不同的是:将步骤(1)中的提取溶媒由体积浓度为95%的乙醇更改为80%的甲醇,提取方式采用加热提取,温度为60℃,提取时间为3h。
[0060]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b。
[0061]
实施例6
[0062]
重复实施例1,不同的是:将步骤(1)中的提取溶媒由体积浓度为95%的乙醇更改为丙酮。
[0063]
对所得产物用nmr及hresims等进行结构分析,确定为目标化合物1a,1b和2a,2b。
[0064]
二、产品的确认
[0065]
实施例1-6所得产物经nmr和hresims等进行结构分析,波谱数据如下所示;并用hplc分析检测,化合物的纯度》95%。
[0066]
化合物1:
[0067]1h nmr(400mhz,cdcl3,25℃)δ
h 7.11(1h,s,h-1),7.63(1h,s,h-4),3.60(1h,d,j=9.4hz,h-6),6.79(1h,s,h-7),7.32(1h,s,h-10),7.68(1h,d,j=8.6hz,h-11),7.50(1h,d,j=8.6hz,h-12),3.20(1h,m,h-1'),1.14(1h,d,j=6.1hz,h-8'),2.70(3h,s,n-me),6.03(2h,s,-och2o-),3.99(3h,s,-och3),3.96(3h,s,-och3);
[0068]
13
c nmr(100mhz,cdcl3,25℃)δ
c 104.7(c-1),147.6(c-2),148.6(c-3),99.6(c-4),131.0(c-4a),138.1(c-4b),71.1(c-6),124.0(c-6a),112.2(c-7),149.1(c-8),148.8(c-9),106.4(c-10),124.0(c-10a),123.7(c-10b),119.6(c-11),124.5(c-12),126.7(c-12a),66.4(c-1'),19.0(c-2'),42.5(n-me),101.2(-och2o-),56.1(-ome),56.2(-ome);
[0069]
hresi-ms m/z 394.1656[m+h]
+
,(calcd for 394.1649)。
[0070]
化合物2:
[0071]1h nmr(400mhz,cdcl3,25℃)δ
h 7.11(1h,s,h-1),7.65(1h,s,h-4),3.69(1h,d,j=9.4hz,h-6),6.86(1h,s,h-7),7.35(1h,s,h-10),7.67(1h,d,j=8.6hz,h-11),7.47(1h,d,j=8.6hz,h-12),3.45(1h,m,h-1'),1.17(1h,d,j=6.1hz,h-8'),2.69(3h,s,n-me),6.06(2h,s,-och2o-),4.00(3h,s,-och3),3.95(3h,s,-och3);
[0072]
13
c nmr(100mhz,cdcl3,25℃)δ
c 100.6(c-1),147.7(c-2),148.4(c-3),104.6(c-4),131.1(c-4a),140.6(c-4b),70.0(c-6),124.9(c-6a),112.3(c-7),149.1(c-8),148.8(c-9),106.6(c-10),124.1(c-10a),123.6(c-10b),119.7(c-11),123.8(c-12),126.8(c-12a),68.9(c-1'),19.7(c-2'),43.6(n-me),101.2(-och2o-),56.3(-ome),56.2(-ome);
[0073]
hresi-ms m/z 394.1658[m+h]
+
,(calcd for 394.1649)。
[0074]
因此,可确定上述化合物1a,1b和2a,2b的结构式如下述式(i)所示:
[0075][0076]
三、产品抗肿瘤作用研究
[0077]
以临床使用的肿瘤治疗药物依托泊苷为阳性对照药,以空白培养基作阴性对照,以snu638(人胃癌细胞),mda-mb-231(人乳腺癌细胞),sk-hep-1(人肝癌细胞),a549(人非小细胞肺癌细胞),hct116(人结肠癌细胞)为受试细胞株用法对化合物进行了体外抗肿瘤活性测试。
[0078]
受试细胞株维持在添加10%胎牛血清和1%抗菌素(aa)的rpmi培养基中(psf;100单位/ml青霉素g钠、100μg/ml链霉素和250ng/ml两性霉素b),细胞在37℃、5%co2、湿润环境下培养。
[0079]
在样品处理72h后使用磺酰罗丹明b(srb)方法测定细胞存活率,每孔加细胞悬液190μl,细胞悬液的细胞浓度为8~3
×
104个/ml。给药组分别加入50μl,10μl,2μl,0.4μl含有不同浓度溶于dmso的本发明化合物的样品10μl,阳性对照组加入20μl,4μl,0.8μl,0.16μl含不同浓度依托泊苷的样品,阴性组不加样品加入和样品组一致的空白dmso 10μl,并进一步孵育72h。用10%三氯乙酸(tca)固定细胞,沉淀剩余的蛋白质,并在0.1%的醋酸中用
srb溶液染色。染色蛋白溶解在10mm tris溶液(ph=10)中,用versamax酶联免疫吸附测定仪在515nm处测定。计算每个给药孔细胞增殖抑制率,并计算其ic
50
值,结果如下述表1所示:
[0080]
表1
[0081][0082]
由表1可知,本发明所述化合物对多种肿瘤细胞均有良好的抑制作用,表明本发明所述化合物具有良好的抗肿瘤作用,有望用于抗肿瘤药物的制备。
[0083]
上述说明是针对本发明较佳可行实施例的详细说明,但实施例并非用以限定本发明的专利申请范围,凡本发明所提示的技术精神下所完成的同等变化或修饰变更,均应属于本发明所涵盖专利范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1