N-氰乙基苯胺的制备方法和设备与流程
n-氰乙基苯胺的制备方法和设备
技术领域
1.本发明涉及一种染料中间体的制备方法,具体涉及一种合成n-氰乙基苯胺的环保方法,属于染料中间体合成领域。
背景技术:
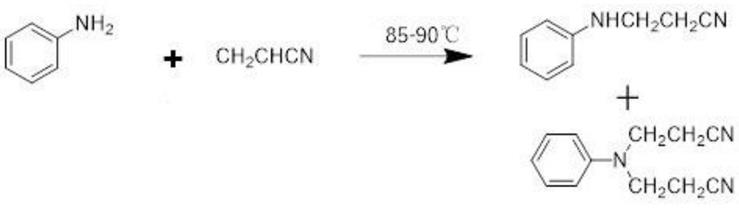
2.n-氰乙基苯胺的结构式为:n-氰乙基苯胺作为一种极为重要的化工原料和中间体,广泛应用于合成染料及一些重要的化工产品领域。例如合成n-氰乙基-n-苄基苯胺,此中间体可用于合成分散橙e-rh。
3.现有的合成n-氰乙基苯胺的路线为:
[0004][0005]
该合成方法中会有n,n-二氰乙基苯胺生成,同时在制备过程中苯胺易氧化。在现有技术中,由于保险粉有很强的还原性,将保险粉溶解在水中,可以防止苯胺被氧化。但是,保险粉在该反应中并未起到很好的效果,苯胺仍然有部分被氧化,影响产品的质量。
[0006]
引用文献1公开一种一步法清洁工艺生产n-氰乙基苯胺和n,n-双氰乙基苯胺,包括如下步骤:反应釜中依次加入苯胺、醋酸、氯化锌、丙烯腈、水及少量对苯二酚;缓慢升温,同时开回馏冷凝器,当回馏温度达到要求后,保持回馏10小时;保温静止1-2小时,将最下层水相装桶分析计量,上面有机相打入中间槽参加下次反应;中间槽保持≥60℃,用连续精馏塔精馏产品,第一出口为轻组分,全部返回反应体系中,主要为hac、h2o、丙烯腈、苯胺,第二出口为n-氰乙基苯胺,第三出口为n,n-双氰乙基苯胺。该种方法的丙烯腈的用量多,且苯胺容易被氧化,因而导致产品纯度低,颜色发黑,团聚在一起,对于后续染料的合成造成影响。
[0007]
引用文献2公开了一种n-氰乙基苯胺零排放生产工艺,是在水相中,加入苯胺、丙烯腈、对苯二酚、催化剂,苯胺、丙烯腈和水的质量比为100:68-75:350-500;催化剂与苯胺的质量比为0.01~0.05:1;对苯二酚与苯胺的质量比为0.002~0.02:1,升温至100~110℃,保持18-22小时后,常压蒸馏回收丙烯腈,然后降温至25℃以下,析出n-氰乙基苯胺,过滤后并用加入苯胺重量的30%-70%水洗涤,得到产品n-氰乙基苯胺;母液水和洗涤水作为下一批n-氰乙基苯胺的生产底水,分析母液水和洗涤水的混合液中催化剂和对苯二酚的含量,补加催化剂和对苯二酚至上述需要量,回收的丙烯腈作为下一批n-氰乙基苯胺的生产的原料,加入丙烯腈和苯胺,进行下一批次生产。该种方法的丙烯腈的用量多,且苯胺容易被氧化,会导致产品纯度低,颜色发黑,团聚在一起,对于后续染料的合成造成影响。
[0008]
引用文献:
[0009]
引用文献1:cn104003905a
[0010]
引用文献2:cn109305929a
技术实现要素:
[0011]
发明要解决的问题
[0012]
鉴于现有技术中存在的技术问题,本发明首先提供一种n-氰乙基苯胺的制备方法。该方法能够使得丙烯腈通过回流再次流入反应体系内,因此,丙烯腈的用量可大大减少,且在基本无氧的环境下进行反应,能够保护苯胺从而避免其被氧化。
[0013]
用于解决问题的方案
[0014]
本发明提供一种n-氰乙基苯胺的制备方法,其包括以下步骤:
[0015]
将催化剂、溶剂和任选地阻聚剂于反应釜中混合后,排出所述反应釜中的氧气,使所述反应釜处于基本无氧的环境;
[0016]
在所述反应釜中,继续添加苯胺和丙烯腈进行保温反应,得到反应产物;
[0017]
在真空条件下,回收所述反应产物中未反应的丙烯腈,得到回收产物;
[0018]
对所述回收产物进行后处理,得到n-氰乙基苯胺。
[0019]
根据本发明所述的制备方法,其中,通过加热放空的方式,以使得所述反应釜处于基本无氧的环境;优选地,所述加热的温度为90~110℃。
[0020]
根据本发明所述的制备方法,其中,所述催化剂包括冰醋酸、盐酸、氯化锌、氯化铝中的一种或两种以上的组合;
[0021]
所述阻聚剂包括对苯二酚;
[0022]
所述溶剂包括水。
[0023]
根据本发明所述的制备方法,其中,所述的苯胺与丙烯腈的投料量的摩尔比为1:1.01~1.25;和/或,所述保温反应的温度为83~95℃,所述保温反应的时间为15~30h。
[0024]
根据本发明所述的制备方法,其中,所述丙烯腈是通过滴加的方式添加至反应体系中的;和/或,所述保温反应的终点为苯胺的纯度低于1.5%。
[0025]
根据本发明所述的制备方法,其中,所述真空条件为0mpa以下,优选为-0.05~-0.07mpa;和/或,回收所述反应产物中未反应的丙烯腈的温度为70~85℃。
[0026]
根据本发明所述的制备方法,其中,以质量计,回收的所述反应产物中未反应的丙烯腈的质量含量为所述丙烯腈的投料量的5~15%。
[0027]
根据本发明所述的制备方法,其中,所述后处理包括利用降温的方式使n-氰乙基苯胺析出的步骤。
[0028]
根据本发明所述的制备方法,其中,通过添加溶剂的方式使后处理体系降温;优选地,降温至后处理体系的温度为30℃以下时,能够使n-氰乙基苯胺析出。
[0029]
根据本发明所述的制备方法,其中,所述n-氰乙基苯胺的收率为90%以上,所述n-氰乙基苯胺的纯度为95%以上,所述n-氰乙基苯胺呈黄色鳞片状。
[0030]
发明的效果
[0031]
本发明的n-氰乙基苯胺的制备方法更为节能环保,产品质量更高。
[0032]
本发明的n-氰乙基苯胺的制备方法减少了丙烯腈用量,并且回收的丙烯腈和水都可以全部套用于下一批生产中,这样能减少大量的消耗与排放。
[0033]
本发明的反应是在无氧体系中进行,无须加保险粉之类的防氧化物质,减少了反应的消耗。
[0034]
另外,本方法制备得到的n-氰乙基苯胺纯度更高,副产物极少,并且晶型呈细鳞片状,比表面积更大,能够更好地作为合成其他产品的原料。
附图说明
[0035]
图1示出了本发明实施例1的n-氰乙基苯胺的合成工艺流程图。
具体实施方式
[0036]
以下,针对本发明的内容进行详细说明。以下所记载的技术特征的说明基于本发明的代表性的实施方案、具体例子而进行,但本发明不限定于这些实施方案、具体例子。
[0037]
另外,为了更好地说明本发明,在下文的具体实施方式中给出了众多的具体细节。本领域技术人员应当理解,没有某些具体细节,本发明同样可以实施。在另外一些实例中,对于本领域技术人员熟知的方法、手段、器材和步骤未作详细描述,以便于凸显本发明的主旨。
[0038]
需要说明的是:
[0039]
本说明书中,使用“数值a~数值b”表示的数值范围是指包含端点数值a、b的范围。
[0040]
如无特殊声明,本发明所使用的单位均为国际标准单位,并且本发明中出现的数值,数值范围,均应当理解为包含了工业生产中所允许的误差。
[0041]
本说明书中,所提及的“一些具体/优选的实施方案”、“另一些具体/优选的实施方案”、“实施方案”等是指所描述的与该实施方案有关的特定要素(例如,特征、性质和/或特性)包括在此处所述的至少一种实施方案中,并且可存在于其它实施方案中或者可不存在于其它实施方案中。另外,应理解,所述要素可以任何合适的方式组合在各种实施方案中。
[0042]
本发明提供一种n-氰乙基苯胺的制备方法,其包括以下步骤:
[0043]
步骤(1)将催化剂、溶剂和任选地阻聚剂于反应釜中混合后,排出所述反应釜中的氧气,使所述反应釜处于基本无氧的环境;
[0044]
步骤(2)在所述反应釜中,继续添加苯胺和丙烯腈进行保温反应,得到反应产物;
[0045]
步骤(3)在真空条件下,回收所述反应产物中未反应的丙烯腈,得到回收产物;
[0046]
步骤(4)对所述回收产物进行后处理,得到n-氰乙基苯胺。
[0047]
本发明减少了约10%的丙烯腈用量,并且回收的丙烯腈和水都可以全部套用于下一批生产中,这样能减少大量的消耗与排放。由于本方法营造了基本无氧的环境体系,无须添加保险粉等之类的防氧化物质,减少了反应的消耗。同时,本方法做出的n-氰乙基苯胺纯度更高,副产物极少,并且晶型呈黄色细鳞片状,比表面积更大,能够更好地作为合成其他产品的原料。
[0048]
步骤(1)
[0049]
本发明通过将催化剂、溶剂和任选地阻聚剂于反应釜中混合后,排出所述反应釜中的氧气,使所述反应釜处于基本无氧的环境;本发明的反应是在基本无氧的环境中进行的,体系的压力小,危险系数小。
[0050]
本发明中所述的“基本无氧”的环境,其含义是在无氧或极少氧含量的环境。
[0051]
在一些具体的实施方式中,本发明可以通过加热放空的方式,以使得所述反应釜处于无氧或极少氧的环境;优选地,所述加热的温度为90~110℃,例如:92℃、95℃、98℃、100℃、102℃、105℃、108℃等。对于加热放空的方式,具体可以在反应釜上设置放空阀,所述放空阀可以是整个反应体系中唯一的出口,通过加热的方式,可以使空气随水蒸气从放空阀中放出,待空气随水蒸气基本放出完全之后关闭放空阀,使整个体系密闭,从而形成釜内基本无氧的环境。
[0052]
本发明中,通过将反应釜内的空气随着水蒸气带出体系外,这样可以使体系处于基本无氧的环境下。在通过加热放空后,既可以除去水中氧气,同时又可以赶走釜内的氧气。
[0053]
进一步,所述反应釜上还可以设置有回流罐,可以将反应釜内混合液升温到一定温度,直到冷凝器(冷凝管)出现回流,保持一定时间后再关闭放空阀,使整个体系密闭。具体地,所述回流的时间可以保持30~60min,这样能将釜内的绝大部分空气赶出体系外。
[0054]
本发明对催化剂、阻聚剂以及溶剂的具体组成不作特别限定,只要可以用于制备n-氰乙基苯胺即可。在一些具体的实施方案中,所述催化剂包括冰醋酸、盐酸、氯化锌、氯化铝中的一种或两种以上的组合;所述阻聚剂包括对苯二酚;所述溶剂包括水。
[0055]
步骤(2)
[0056]
在所述反应釜中,继续添加苯胺和丙烯腈进行保温反应,得到反应产物。可以先将苯胺添加至反应釜中,再将丙烯腈添加至反应釜中。
[0057]
在一些具体的实施方案中,将苯胺添加至反应釜后,待反应釜内自然降到一定温度时慢慢将丙烯腈添加至反应釜内。通过缓慢的将丙烯腈添加至反应釜内,可以使丙烯腈与苯胺在无氧的环境下充分混合,反应更加彻底。
[0058]
在本发明中,所述丙烯腈是通过滴加的方式添加至反应体系中的。具体地,将苯胺添加至反应釜内后,可以开循环水降温至83~90℃,例如84℃、85℃、86℃、87℃、88℃、89℃等开始滴加丙烯腈,滴加过程有反应热产生,滴加速度不宜过快,使体系维持在一定温度下。优选地,本发明所述丙烯腈的滴加时间可以为3~6h,例如:3.2小时、3.5小时、3.8小时、4小时、4.2小时、4.5小时、4.8小时、5小时、5.2小时、5.4小时、5.8小时等。
[0059]
具体地,所述的苯胺与丙烯腈的投料量摩尔比为1:1.01~1.25,例如:1:1.05、1:1.08、1:1.1、1:1.12、1:1.15、1:1.18、1:1.2、1:1.22等;和/或,所述保温反应的温度为83~95℃,例如:84℃、85℃、86℃、87℃、88℃、89℃、90℃、91℃、92℃、93℃、94℃等;所述保温反应的时间为15~30h,例如:18h、20h、22h、25h、28h等。本发明的方法的保温反应过程中,温度和时间有着很大的影响,温度过高、反应时间过长都会导致反应产生副产物,影响产品的质量。
[0060]
另外,在本发明中,所述保温反应的终点为苯胺的纯度低于1.5%,优选为hplc检测苯胺的纯度低于1.5%。具体可以通过保温一段时间后取样,若苯胺的纯度高于1.5%则继续保温1~10h,直至苯胺的纯度低于1.5%,即可判定反应到终点。
[0061]
步骤(3)
[0062]
待反应达到终点之后,在真空条件下,回收所述反应产物中未反应的丙烯腈,得到回收产物。具体地,所述反应结束后,还存在一定量未反应的丙烯腈,通过在真空条件下对其进行回收后还可以用作下一批生产中。所述回收产物为除去未反应的丙烯腈后剩余的物
质。
[0063]
具体地,所述真空条件为0mpa以下,优选为-0.05~-0.07mpa,例如:-0.01mpa、-0.03mpa、-0.06mpa、-0.08mpa等;和/或,回收所述反应产物中未反应的丙烯腈的温度为70~85℃,例如:72℃、73℃、74℃、75℃、76℃、78℃、80℃、81℃、82℃、83℃、84℃等。
[0064]
另外,以质量计,回收的所述反应产物中未反应的丙烯腈的质量含量为所述丙烯腈的投料量的5~15%,例如6%、7%、8%、9%、10%、11%、12%、13%、14%等。
[0065]
步骤(4)
[0066]
进一步,通过对所述回收产物进行后处理,从而得到n-氰乙基苯胺。
[0067]
在一些具体的实施方案中,所述后处理包括利用降温的方式使n-氰乙基苯胺析出的步骤。具体地,可以通过在反应釜中添加溶剂的方式以达到降温的效果。本步骤中的溶剂可以为水等常用溶剂。
[0068]
进一步,可以在反应釜中先添加一部分的溶剂,使温度降至38~42℃后,再缓慢降低体系的温度。具体可降温至后处理体系的温度为30℃以下时,能够使n-氰乙基苯胺析出。一般而言,所述n-氰乙基苯胺的最低溶解温度为25℃左右,因此,当后处理体系的温度为30℃以下回收产物中会有n-氰乙基苯胺析出。一般当后处理体系的温度为20~30℃时,回收产物中的n-氰乙基苯胺可以完全析出。
[0069]
另外,对于溶剂的加入量,本发明不作特别限定,可以根据需要进行添加,具体地,溶剂的添加量可以是步骤(1)中溶剂的添加量的1~3倍,例如:1.2倍、1.5倍、1.8倍、2倍、2.2倍、2.5倍、2.8倍等。
[0070]
进一步,在本发明中,所述n-氰乙基苯胺的收率为90%以上,所述n-氰乙基苯胺的纯度为95%以上,所述n-氰乙基苯胺呈黄色鳞片状。
[0071]
本发明的制备方法更为节能环保,产品质量更高。本发明的制备方法减少了约10%的丙烯腈用量,并且回收的丙烯腈和水都可以全部套用于下一批生产中,这样能减少大量的消耗与排放。此外,由于本发明的制备方法使反应在无氧条件下进行,无须加保险粉之类的防氧化物质,减少了反应的消耗。同时,本方法做出的n-氰乙基苯胺纯度更高,副产物极少,并且晶型呈细鳞片状,比表面积更大,能够更好地作为合成其他产品的原料。
[0072]
实施例
[0073]
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市购获得的常规产品。
[0074]
实施例1
[0075]
如图1所示,在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在88℃,保温16h后使用hplc检测(苯胺:1.19%,n-氰乙基苯胺:95.81%),反应达到终点,开始抽真空至-0.06mpa,温度保持在80℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。回收毕,加入工业水
1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为97.39%,收率为94.75%,产品呈亮黄色鳞片状。
[0076]
实施例2
[0077]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将246g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.20),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在88℃,保温16h后使用hplc检测(苯胺:1.24%,n-氰乙基苯胺:96.11%),反应达到终点,开始抽真空至-0.06mpa,温度保持在80℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈24g,水306g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为97.59%,收率为94.58%,产品呈亮黄色鳞片状。
[0078]
实施例3
[0079]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺打入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将215g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.05),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在88℃,保温16h后使用hplc检测(苯胺:1.55%,n-氰乙基苯胺:94.82%),反应未达到终点,继续保温2h后使用hplc检测(苯胺:1.47%,n-氰乙基苯胺:94.86%),反应达到终点,开始抽真空,回收丙烯腈至-0.06mpa,温度保持在80℃,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈21g,水309g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为96.44%,收率为93.15%,产品呈亮黄色鳞片状。
[0080]
实施例4
[0081]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在83℃,约5h滴完丙烯腈。釜内温度控制在85℃,保温16h后使用hplc检测(苯胺:1.91%,n-氰乙基苯胺:93.72%),反应未达到终点,继续保温8h后使用hplc检测(苯胺:1.44%,n-氰乙基苯胺:95.15%),反应达到终点,开始抽真空至-0.06mpa,温度保持在80℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为96.74%,收率为94.34%,产品呈亮黄色鳞片状。
[0082]
实施例5
[0083]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在95℃,保温16h后使用hplc检测(苯胺:0.97%,n-氰乙基苯胺:95.12%),反应达到终点,开始抽真空至-0.06mpa,温度保持在80℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为96.37%,收率为92.90%,产品呈暗黄色鳞片状。
[0084]
实施例6
[0085]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在90℃,保温16h后使用hplc检测(苯胺:1.17%,n-氰乙基苯胺:95.79%),反应达到终点,开始抽真空至-0.05mpa,温度保持在75℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到30℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为97.42%,收率为93.63%,产品呈亮黄色鳞片状。
[0086]
实施例7
[0087]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g。投毕,升温至100℃,开启回流罐的冷凝水,同时开启回流罐上方的放空阀。保持回流30min后关闭放空阀,将360g苯胺加入釜内,过程中保持釜内密闭。此时釜内温度为85℃左右,再用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在85℃,约5h滴完丙烯腈。釜内温度控制在88℃,保温16h后使用hplc检测(苯胺:1.14%,n-氰乙基苯胺:95.98%),反应达到终点,开始抽真空至-0.07mpa,温度保持在85℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到20℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为97.28%,收率为94.88%,产品呈亮黄色鳞片状。
[0088]
对比例1
[0089]
在2l高压釜中加入底水560g、对苯二酚2.96g、氯化锌36g、冰醋酸36g、保险粉1.44g。投毕,升温至75℃,将360g苯胺加入釜内,过程中保持釜内密闭。用泵将225g丙烯腈慢慢滴入釜内(苯胺与丙烯腈的摩尔比为1:1.10),过程中控制温度在85℃以下,且注意釜内压力小于0.1mpa,约5h滴完丙烯腈。釜内温度控制在88℃,保温16h后使用hplc检测(苯胺:1.33%,n-氰乙基苯胺:94.89%),反应达到终点,开始抽真空至-0.06mpa,温度保持在80℃回收丙烯腈,此回收过程回收到水和丙烯腈的混合物约330g,其中丙烯腈22g,水308g。
回收毕,加入工业水1l,开启釜内循环水,当釜内温度降到40℃左右,减小循环水量,使釜内温度慢慢降到25℃,此时n-氰乙基苯胺完全析出,抽滤,洗涤。最终得到产品n-氰乙基苯胺,hplc检测纯度为95.57%,收率为94.52%,产品中有小部分呈黑色球状。
[0090]
实施例1与对比例1相比可知,对比例1的苯胺被部分氧化,导致产品有部分呈球状。可见,采用本技术的方法,不仅能提高产品的纯度,而且能得到外观更为合格的产品。
[0091]
对比例2
[0092]
在2l反应釜中加入360g苯胺,16g氯化锌,0.72g对苯二酚,密闭反应釜,开冷凝水和真空,吸入30g冰醋酸,31g水,随后再加入246g丙烯腈,慢慢升温,70℃左右有回馏,回馏2.5小时左右后,温度在90℃左右,保持90-100℃回馏12小时左右,取样中控,直至hplc检测苯胺的含量为0.1
±
0.02%,n,n-双氰乙基苯胺10
±
2%,n-氰乙基苯胺和n,n-双氰乙基苯胺加和大于98%。终点到后静置2小时,分掉水相,取有机相进行连续精馏,第一出口为轻组分,全部返回反应体系中,第二出口为n-氰乙基苯胺,第三出口为n,n-双氰乙基苯胺。产品n-氰乙基苯胺,hplc检测纯度为89.23%,收率为88.64%。
[0093]
从对比例2可以看出,其相关产品的纯度和收率都不高。
[0094]
对比例3
[0095]
在2l高压釜中,依次加水1420g、30%盐酸7.7g、对苯二酚2.6g、苯胺360g、丙烯腈244g,慢慢升温至110℃保持20小时后,常压蒸馏回收过量的丙烯腈,反应物料慢慢降温至25℃,真空过滤并用少量水洗涤得到产品n-氰乙基苯胺,hplc检测纯度为91.15%,收率为92.30%,产品呈黑色球状。
[0096]
从对比例3可以看出,苯胺被部分氧化,导致产品呈球状,且产品纯度和收率不高。
[0097]
本发明的上述实施例仅仅是为清楚地说明本发明所作的举例,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动。这里无需也无法对所有的实施方式予以穷举。凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明权利要求的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1