一种DeepVent(exo-)DNA聚合酶的制备方法与流程
一种deep vent(exo-)dna聚合酶的制备方法
技术领域
1.本发明涉及dna聚合酶技术领域,具体为deep vent(exo-)dna聚合酶的制备方法。
背景技术:
2.deep vent(exo-)dna聚合酶是一种高保真的耐热dna聚合酶,具有高保真性的原因,部分是由于其自身具有3'
→
5'核酸外切酶校读活性。deep vent dna聚合酶比vent dna聚合酶在95℃到100℃之间更稳定,其在100℃的半衰期是8小时。deep vent(exo-)dna聚合酶是利用基因工程技术去除了deep vent dna聚合酶的3'
→
5'核酸外切酶校读活性而得。
3.cn 101255435 a公开了耐热性dna聚合酶(bcady-pol)基因及其编码蛋白的应用,采用ni离子柱进行纯化,得到的蛋白纯度仍然无法满足二代测序的要求。
4.cn 104560908 a公开了一种重组大肠杆菌taq dna聚合酶的纯化方法,包括以下步骤:(1)破菌;(2)热处理;(3)盐析;(4)ni离子金属螯合亲和层析;(5)透析;(6)制剂分装。然而该方法得到的taq dna聚合酶的热稳定性较差。
5.cn 108265039 a公开了一种突变taq dna聚合酶及其纯化方法,将天然taq dna聚合酶进行突变,去除了其5
’‑3’
的dna外切酶活性,并且引入e507r,e742a和a743h(氨基酸位置根据野生型taq dna聚合酶所定)突变。然而所述taq dna聚合酶的热稳定性较差。
6.cn 110938611 a公开了一种突变deep sea dna聚合酶及其纯化方法,将野生型dna聚合酶的多位点氨基酸进行突变,得到的deep sea dna聚合酶去除了野生型dna聚合酶的部分3
’→5’
核酸外切酶校读活性。然而该方法容易产生大肠杆菌基因组和二价金属离子残留,在二代测序使用过程中的结果往往不理想。
技术实现要素:
7.基于上述情况,鉴于以上的问题,本发明提供一种deep vent(exo-)dna聚合酶及其制备方法,该方法制备的dna聚合酶热稳定性好、纯度高、金属离子及核酸残留率低,在二代测序中反馈良好,无抑制。
8.为了解决上述技术问题,本发明提供如下技术方案:
9.一种deep vent(exo-)dna聚合酶的制备方法,包括以下步骤:
10.(1)设计含his标签基因的deep vent(exo-)dna聚合酶表达基因,通过大肠杆菌表达系统表达获得菌体;
11.(2)裂解菌体后获得粗产物;
12.(3)对粗产物加入终浓度为0.1%~0.5%的pei进行预处理;
13.(4)蛋白纯化过程包括依次处理的疏水层析、ni亲和层析、阳离子交换层析、凝胶过滤层析、chelex亲和层析,最后获得纯化的deep vent(exo-)dna聚合酶。
14.优选的,对所述步骤(2)获得的粗产物进行热变性处理,处理条件为温度70~90℃,加热时间30~60分钟。
15.优选的,所述步骤(4)中的疏水层析的结合缓冲液电导为55~135ms/cm。
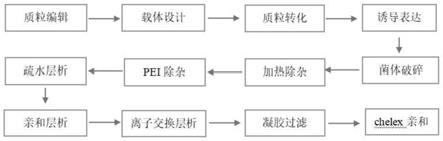
16.优选的,所述步骤(4)中的疏水层析的洗脱缓冲液成分,20mm pb,0~1.2m(nh4)2so4,洗脱梯度,0~100%,共分为5个洗脱步骤。
17.优选的,所述步骤(4)中的疏水层析的结合缓冲液组分为:20mm pb+1m硫酸铵,ph=7.6,25℃,所述步骤(4)中的疏水层析的洗脱缓冲液成分为20mm pb,ph=7.6,25℃。
18.优选的,所述步骤(4)中的chelex亲和层析的2倍的洗脱缓冲液具体成分为:20mm tris-hcl,200mmkcl,2mm dtt,0.2mm edta,0.2%tween20,ph:7.4。
19.本发明的技术原理如下:
20.蛋白纯化过程包括依次处理的疏水层析、ni亲和层析、阳离子交换层析、凝胶过滤层析、chelex亲和层析。
21.第一步疏水层析——因为在预处理阶段加入了pei,而pei带正电,会和树脂上的ni2+形成竞争吸附,导致树脂脱ni2+,降低纯化效果,所以不能直接使用ni柱亲和层析,不用离子交换层析的原因在于破碎液有较高的盐浓度,使用离子交换层析就必须对破碎液进行稀释,这会大量增加上样体积,拖长整个纯化过程所需要的时间;
22.第二步ni柱亲和层析——因为在设计载体时加入了his标签,使得ni柱亲和层析是最高效的纯化方式,而破碎液中加入了pei,故不放在第一步;
23.第三步离子交换层析——因为经过ni柱亲和层析之后,到这步时的上样体积已经较少,且ni柱洗脱液的电导相比之前的也较低。
24.第四步、第五步的层析缓冲液为2
×
洗脱缓冲液,使用时稀释至1倍。
25.与现有技术相比,本发明所达到的有益效果是:
26.本发明通过往裂解菌体后得到的粗产物中加入pei的方法除去部分核酸,并采用疏水层析、ni亲和层析、离子交换层析、凝胶过滤层析和chelex亲和层析的纯化方法,除去剩余的核酸残留以及二价金属离子残留,减少了核酸残留和二价金属离子对实验的负面影响,提高了二代测序建库的效果。
附图说明
27.附图用来提供对本发明的进一步理解,并且构成说明书的一部分,与本发明的实施例一起用于解释本发明,并不构成对本发明的限制。在附图中:
28.图1为dna聚合酶的纯化方法流程图;
29.图2为不同电导下deep vent(exo-)dna的sds-page电泳图;
30.图3为核酸残留对比图;
31.图4为二价金属残留对比图;
32.图5为deep vent(exo-)dna功能检测图;
33.图6为高gc含量模板的pcr扩增图。
具体实施方式
34.下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
35.实施例中自产的deep vent(exo-)为通过本发明方法获得的deep vent(exo-)dna聚合酶,a公司的deep vent(exo-)dna聚合酶提供产品和货号为:厂家:neb,货号:m0258s,批号:10049672。
36.实施例:
37.1.蛋白纯化:
38.(1)质粒转化,取1支rosetta感受态,加入1~4μg deep vent(exo-)质粒,热激90s,加入1000μl新鲜无抗lb液体培养基,置于摇床200rpm、37℃培养1h;取200μl菌液均匀涂布于kan+抗性的lb平板中(0.05~0.5%),37℃恒温培养箱过夜培养;
39.(2)deep vent(exo-)诱导表达,挑取长势正常的单克隆菌落到150ml kan+抗性的lb液体培养基中(100μg/ml kan+),置于摇床200rpm、37℃过夜培养。取10ml培养好的菌种接种于1l的kan+抗性的lb液体培养基中(100~400μg/mlkan+),200rpm 37℃培养摇床培养4h左右。使用可见分光光度计监测od600值,待培养的菌液浓度od600在0.8-1.0左右时加入iptg终浓度为1.0~4.0mm,温度控制在16~37℃,开始计时,过夜诱导表达。使用落地离心机收菌,设置6500rpm,4℃,15min,去尽残留培养基收集沉淀,称量所有菌体湿重并记录,将所有菌体保存在-80℃条件下;
40.(3)高压破碎,使用电子天平称量deep vent(exo-)dna聚合酶湿菌20
±
1g,放入250ml烧杯中;用量筒量取200
±
10ml裂解buffer a(湿菌:buffer比例为1g:5~15ml)加入烧杯中,使用涡旋混匀仪快速涡旋均匀直至肉眼观察无明显菌块,磁力搅拌器搅拌至无块状菌体。依次开启高压均质机制冷电源、制冷、循环按钮,并设置制冷温度:4℃。高压细胞破碎仪冷阱提前循环预冷至4℃。和菌体处理开启高压均质机电源,用1000
±
100ml工艺用水清洗高压均质机管道,设置参数:速度50、压力0-20bar,排尽管路中75%乙醇。然后加入500ml工艺用水清洗管路,然后加入200ml裂解buffer a润洗管路后排空,将重悬菌液缓慢倒入高压均质机进样杯中,设置参数:速度50、压力800-1000bar;开始破碎,接收破碎后液体,破碎6次,细胞破碎液置于冰浴暂存。破碎完毕后,先用2000
±
100ml工艺用水清洗高压均质机管道,再用300
±
100ml 75%乙醇清洗高压均质机管道,并保持管道内充满75%乙醇。关闭高压均质机电源,关闭高压均质机制冷系统电源;
41.(4)破碎液离心离心:将破碎液转移至离心管中,两两配平,15000rpm、4℃离心30min。分离沉淀和上清;
42.(5)热变性,设置数显恒温水浴锅温度,转移细胞破碎液至玻璃烧杯中,并将其置入水浴锅中,置顶式搅拌器搅拌加热,菌液温度达到70~90℃时开始计时,总体加热时间30~60min;
43.(6)pei沉淀,将离心后的上清液,边磁力搅拌边加入10%pei母液(ph:6~8),直至加入的pei终浓度为0.1%~0.5%为止,室温磁力搅拌平衡10min,然后置于4℃放置20min;15000rpm,4℃,离心30min;
44.(7)破碎液处理离心,将热变性后液体置于冰上降温约4℃后,转移至50ml离心管中,用电子天平两两配平,当离心管为奇数时,使用空白管子加工艺用水配平,15000rpm、4℃离心30min。过滤:将上清转移至干净烧杯中,依次分别用0.45μm和0.22μm滤膜过滤,2~8℃待用时间不超过30min;超过待用时间,需重新用0.22μm滤膜过滤,然后使用;
45.(8)加入3m(nh4)2so4补盐,使用电导率仪测试上清,记录电导率。加入3m(nh4)
2so4母液,边磁力搅拌边加入(nh4)2so4(少量多次加入上清中),直至电导显示为55~135ms/cm左右;
46.(9)疏水层析,结合缓冲液电导为55~135ms/cm,洗脱缓冲液关键成分,20mm pb,0~1.2m(nh4)2so4,洗脱梯度,0~100%,共分为5个洗脱步骤;
47.具体为:结合缓冲液组分为:20mm pb+1m硫酸铵,ph=7.6,25℃;洗脱缓冲液成分为20mm pb,ph=7.6,25℃;
48.具体洗脱步骤见下表:表中deep vent(exo-)butyl buffer a为结合缓冲液,deep vent(exo-)butyl buffer b为洗脱缓冲液。
[0049][0050]
(10)亲和层析,elution buffer关键成分,10~30mm tris-hcl,100~300mm nacl,35~350mm咪唑,洗脱梯度,0~100%,共分为5个洗脱步骤;
[0051]
(11)离子交换层析,elution buffer关键成分,10~30mm tris-hcl,25~500mm nacl,洗脱梯度,0~100%,共分为5个洗脱步骤;
[0052]
(12)凝胶过滤层析,20mm tris-hcl,200mm kcl,2mm dtt,0.2mm edta,0.2%tween20;
[0053]
(13)chelex亲和,20mm tris-hcl,200mm kcl,2mm dtt,0.2mm edta,0.2%tween20;
[0054]
(14)各步骤流程图如上图1所示,图中pei除杂、疏水层析和chexel亲和为新增步骤;
[0055]
2.根据sds-page电泳展现的结果发现,不同盐浓度对deep vent(exo-)dna有着不同程度的影响,具体如上图2所示;
[0056]
3.利用qpcr对纯化后的蛋白做核酸残留检测时,ⅰ为加入pei和疏水层析后的新工艺,其在45个循环时依旧没有起峰,ⅱ为没有刻意除核酸的旧有工艺,其在36个循环之后出现起跳,证明其存在些许核酸残留,具体如上图3所示。
[0057]
4.在氨水作为缓冲溶液的条件下,以丁二酮肟作为显色剂,过硫酸铵作为氧化剂,用分光光度法在435nm的波长下,测定镍离子含量的相对大小。结果显示,chelex层析柱能明显螯合ni柱亲和后剥离的ni2+,具体如上图4所示;
[0058]
5.在使用通用模板进行功能检测时,a公司和自产的deep vent(exo-)的功能大致
相当,自产的deep vent(exo-)表现相对更好些,结果如上图5所示(1/4~1/128为聚合酶的稀释梯度);
[0059]
6.在使用高gc模板进行功能检测时,a公司和自产的deep vent(exo-)的功能大致相当,自产的deep vent(exo-)在低浓度时表现相对更好些,结果如上图6所示(1/4~1/128为聚合酶的稀释梯度);
[0060]
7.将纯化好的酶进行二代测序,新工艺在去除二价金属离子后,文库cdna浓度明显高于未去除二价金属离子的旧工艺,结果如下表所示。
[0061][0062]
需要说明的是,在本文中,诸如第一和第二等之类的关系术语仅仅用来将一个实体或者操作与另一个实体或操作区分开来,而不一定要求或者暗示这些实体或操作之间存在任何这种实际的关系或者顺序。而且,术语“包括”、“包含”或者其任何其他变体意在涵盖非排他性的包含,从而使得包括一系列要素的过程、方法、物品或者设备不仅包括那些要素,而且还包括没有明确列出的其他要素,或者是还包括为这种过程、方法、物品或者设备所固有的要素。
[0063]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1