一种MAC型多簇串联线性半抗原、人工抗原及其制备方法和应用
一种mac型多簇串联线性半抗原、人工抗原及其制备方法和应用
技术领域
1.本发明涉及抗原抗体检测技术领域,更具体地,涉及一种含有3-甲基-喹恶啉-2-羧酸(mqca)、金刚烷胺(amd)及氯霉素胺(capa)特征骨架结构的 mac型多簇串联线性半抗原、人工抗原及其制备方法和应用。
背景技术:
2.目前由于抗生素等兽药的滥用,造成畜禽产品安全问题依然比较严重。其中金刚烷胺、喹乙醇、氯霉素等禁用抗生素违规滥用现象较为普遍,对消费者身心健康造成极大潜在危害。目前,畜产品兽药残留分析主要大型精密仪器确证技术与免疫分析筛查技术相结合模式进行,其中仪器确证技术方法主要为色谱-质谱联用技术,但存在样品预处理操作复杂、费时、成本高,不适合现场检测等缺点。免疫分析法具有灵敏度与常规的仪器分析一致,适合现场筛选,简单、快速、成本低、前处理简单等优点。但是,目前食品中农兽药残留检测以单残留免疫检测方法为主,例如中国专利中分别公开了有单独针对金刚烷胺、喹乙醇、氯霉素的免疫检测方法。然而鉴于兽药种类的多样性及其复配使用的广泛性和复杂性,使兽药多残留问题非常突出,仅发展单残留免疫检测技术已不能满足实际检测需求,因此,如何实现对非共有结构抗生素多残留免疫检测是一种新的发展方向。但是目前缺少用于同时检测非共有结构小分子药物金刚烷胺、喹乙醇、氯霉素的相关半抗原、抗原和抗体。
技术实现要素:
3.本发明为了克服现有技术中存在的上述缺陷和不足,提供一种可以实现非共有结构有机小分子药物识别的mac型多簇串联线性半抗原。通过将喹乙醇、金刚烷胺、氯霉素3种抗生素药物分子骨架结构单元通过适当线性偶联间隔臂链接成多簇串联线性半抗原。
4.本发明的第二个目的是提供所述mac型多簇串联线性半抗原的制备方法。
5.本发明的第三个目的是提供所述mac型多簇串联线性半抗原的应用。
6.本发明的第四个目的是提供一种mac型多簇串联线性人工抗原。
7.本发明的第五个目的在于提供所述mac型多簇串联线性人工抗原的制备方法。
8.本发明的第六个目的在于提供所述mac型多簇串联线性人工抗原的应用。
9.本发明的第七个目的在于提供一种多簇串联线性抗原特异性抗体。
10.本发明的上述目通过以下技术方案予以实现:
11.一种含3-甲基-喹恶啉-2-羧酸(mqca)、金刚烷胺(amd)及氯霉素胺(capa) 骨架结构单元的mac型多簇串联线性半抗原,具有式ⅰ所示分子结构:
[0012][0013]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即所述r1和r2分别选自乙基至戊基中的任意一种。
[0014]
本发明的mac型多簇串联线性半抗原是通过将金刚烷胺(amd)、喹乙醇代谢物(mqca)和氯霉素(capa)的骨架结构单元通过不同间隔臂的氨基酸树枝状分子连接制成。
[0015]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0016]
进一步优选地,所述mac型多簇串联线性半抗原结构式如下mac-4c或 mac-6c所示:
[0017][0018]
所述mac型多簇串联线性半抗原的制备方法,包括以下步骤:
[0019]
s1.分别制备喹乙醇代谢物、金刚烷胺、氯霉素中间体:以乙酰乙酸甲酯为原料经过nbs溴代、4-硝基邻苯二胺环化、还原硝基、上氨基保护基、酯键水解,得到式ii所示喹乙醇代谢物中间体;以金刚烷为原料经过硝化、硝基还原、氨基酰化、水解后在其中1个氨基上连接叔丁氧羰基保护的氨基酸手臂,然后,在另1个氨基上连接芴甲氧羰酰基,最后脱除boc保护基,得到式iii所示金刚烷胺中间体;将氯霉素的二氯乙酰基水解除去,再通过酰化缩合反应偶联得到式 iv所示氯霉素中间体;
[0020][0021]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即所述r1和r2分别选自乙基至戊
基中的任意一种;
[0022]
s2.将式iii所示金刚烷胺中间体通过其氨基分别与式ii所示喹乙醇代谢物中间体的羧基缩合偶联,然后脱除金刚烷胺中间体氨基上的fmoc保护基后,将氨基与式iv所示氯霉素中间体的羧基缩合偶联得到中间体,然后再脱除喹乙醇代谢物中间体端氨基上tmb保护基后,得到式ⅰ所示mac型多簇串联线性半抗原。
[0023]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0024]
本发明还提供所述mac型多簇串联线性半抗原在制备mac型多簇串联线性人工抗原中的应用。
[0025]
一种mac型多簇串联线性人工抗原,通过式ⅰ所示半抗原的氨基偶联载体蛋白得到,具有式v所示分子结构:
[0026][0027]
其中,r1和r2分别选自碳原子个数为2~5的烷基,即所述r1和r2分别选自乙基至戊基中的任意一种。
[0028]
优选地,所述r1选自丙基或戊基,r2选自乙基或丁基。
[0029]
进一步优选地,所述mac型多簇串联线性人工抗原结构式如下式viii或式 ix所示:
[0030][0031]
优选地,所述载体蛋白为乳铁蛋白(lf)、钥孔血蓝蛋白(klh)或卵清蛋白(ova),其中lf、klh用于制备免疫原,ova用于制备包被原。
[0032]
所述mac型多簇串联线性人工抗原的制备方法,在式ⅰ所示mac型多簇串联线性半抗原的氨基上偶联载体蛋白得到。
[0033]
优选地,所述偶联为通过重氮化法。
[0034]
作为一种优选地可实施方式,所述mac型多簇串联线性人工抗原的制备方法,包括如下步骤:将式ⅰ所示mac型多簇串联线性半抗原溶于2ml水中,用 0.1m hcl调ph至2.0,加入10mg/ml nano2约350μl,4℃避光反应30min,记为a液。称取载体蛋白10mg溶于4ml cbs缓
冲溶液(0.01m,ph 9.0)中,搅拌溶解制成b液。磁力搅拌下,吸取a液逐滴加入到b液中,4℃下磁力搅拌反应12h。将反应液于4℃下用pbs透析3天,每天换2次透析液。即可得到免疫原,冻存于-20℃冰箱中,备用。
[0035]
本发明还提供所述mac型多簇串联线人工抗原在制备制备单/多克隆抗体、单链抗体或纳米抗体中的应用。
[0036]
一种多克隆抗体,是以式v所示mac型多簇串联线性人工抗原免疫小鼠制备得到。
[0037]
优选地,所述多克隆抗体是以载体蛋白为乳铁蛋白(lf)或钥孔血蓝蛋白 (klh)的mac型多簇串联线人工抗原为免疫原,选取适龄雌性balb/c小鼠进行免疫,第4次免疫和第5次免疫后测定抗血清的效价和抑制率,待性能稳定后收集小鼠血清,得到多克隆抗体。
[0038]
本发明还提供一种同时检测金刚烷胺、喹乙醇、氯霉素的免疫检测方法,采用间接elisa,以上述任一所述人工抗原为包被原,采用上述所述多克隆抗体对待测样品进行检测。
[0039]
本发明具有以下有益效果:
[0040]
本发明提供了一种mac型多簇串联线性半抗原,其结构通式如式i所示,将金刚烷胺(amd)、喹乙醇代谢物(mqca)和氯霉素胺(capa)3种药物骨架结构分别采用不同长短的氨基烷基羧酸或烷基二酸依次连接形成多簇串联线性半抗原,基于该半抗原偶联载体蛋白制备获得人工抗原,再免疫动物测试其免疫应答效果,诱导获得的多抗血清在1μg/ml包被抗原下效价在8~16k之间,对1μg/ml mac型多簇串联线性半抗原抑制率在51~85%之间,表明本发明制备的多簇串联线性半抗原及其人工抗原具有良好的免疫原性,可以用于后续制备单克隆抗体、单链抗体和纳米抗体。本发明同时为丰富小分子有机物多组分识别核心试剂抗体提供新思路,而且为创新抗生素多残留分析方法奠定基础,具有良好的应用前景。
附图说明
[0041]
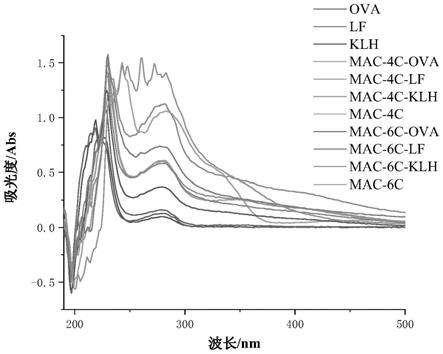
图1为mac型多簇串联线性半抗原、人工抗原、载体蛋白紫外扫描图谱。
具体实施方式
[0042]
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。除非特别说明,以下实施例所用试剂和材料均为市购。
[0043]
实施例1mac型多簇串联线性半抗原的制备
[0044]
1、一种mac-4c半抗原的制备方法,包括如下步骤:
[0045]
s1.中间体3的制备
[0046][0047]
将乙酰乙酸甲酯即化合物1(50g,431mmol)于水(1l)中,在70℃下加入n-溴代琥珀酰亚胺(即nbs)(88g,494mmol)并在室温下搅拌半小时至体系透明。随后加入4-硝基邻苯二胺即化合物2(63g,412mmol)并在70℃下继续搅拌8小时,经液-质联用质谱仪(即lc-ms)监
nmr(400mhz,dmso)δ7.88(d,j =7.5hz,1h),7.71(d,j=7.4hz,1h),7.48
–
7.36(m,1h),7.33(td,j=7.4,1.1hz, 1h),7.11(s,1h),6.76(s,1h),4.18(s,1h),2.94
–
2.79(m,1h),2.11(s,2h),2.03
–ꢀ
1.93(m,3h),1.92
–
1.61(m,4h),1.54(dt,j=14.3,7.2hz,2h),1.37(s,5h)。
[0066]
s8:中间体12的制备
[0067][0068]
将中间体11(6.9g,12mmol)溶于4m的盐酸二氧六环溶液中(50ml) 中并在室温下继续搅拌反应1小时。薄层色谱(tlc)显示反应完全。将反应液浓缩得中间体12(7g)的淡黄色油状液体。ms(esi)m/z(m+h)
+
474.2。1h nmr (400mhz,dmso)δ7.89(d,j=7.5hz,6h),7.71(d,j=7.4hz,2h),7.52(s,1h), 7.41(t,j=7.2hz,3h),7.33(td,j=7.4,1.1hz,2h),7.12(s,1h),4.20(d,j=14.9 hz,3h),2.74(dd,j=14.1,6.4hz,2h),2.17
–
2.03(m,6h),1.78(ddd,j=25.2, 22.1,15.0hz,12h),1.51(s,2h)。
[0069]
s9.中间体13的制备
[0070][0071]
将中间体6(3.5g,9.9mmol)、2-(7-氮杂苯并三氮唑)-n,n,n',n'-四甲基脲六氟磷酸酯(即hatu)(4.5g,11.8mmol)和n,n-二异丙基乙胺(即diea)(3.8 g,30mmol)溶于20ml二氯甲烷中,并在室温下继续搅拌反应1小时。将上述液体加入正在搅拌的中间体12(6g)的dcm(100ml)溶液中,并在室温下继续搅拌反应半小时,经液-质联用质谱仪(即lc-ms)监测反应完全后,加入水(100ml),用dcm(100ml
×
3)萃取,有机层用饱和食盐水洗涤(100ml) 后用无水硫酸钠干燥过滤后浓缩。剩余物经柱层析(pe:ea=100:0~0:100)分离纯化得中间体13(6.9g,收率:85.9%,8.5mmol)的橙色固体。ms(esi)m/z(m+h) +
809.2。1h nmr(400mhz,cdcl3)δ8.24(t,j=6.1hz,1h),7.81
–
7.70(m,3h), 7.56(t,j=12.1hz,2h),7.39(t,j=7.3hz,2h),7.34
–
7.28(m,2h),7.24(d,j=8.3 hz,1h),7.18(dd,j=9.1,2.6hz,1h),6.96(d,j=2.5hz,1h),6.51(d,j=2.3hz, 1h),6.46(dd,j=8.2,2.3hz,1h),4.38(d,j=5.5hz,2h),4.19(t,j=6.1hz,1h), 3.84(d,j=11.5hz,3h),3.80(s,3h),3.54(q,j=6.5hz,2h),3.14(dt,j=12.8,6.4 hz,1h),3.04(d,j=8.3hz,3h),2.80(s,6h),2.21(t,j=6.9hz,4h),1.95(dt,j= 30.2,8.9hz,9h),1.45(t,j=5.6hz,6h)。
[0072]
s10.中间体15的制备
[0073]
[0074]
将氯霉素即化合物14(25g,77.6mmol)于hcl(1m,500ml)中,在100℃下搅拌过夜。lc-ms显示反应完全。将反应液冷却至室温,用乙酸乙酯(ea) (500ml
×
3)萃取除掉二氯乙酸。剩余水相旋干得氯霉素水解物——氯霉素胺 (capa)即中间体15(15g,收率:91.1%,70.7mmol)的白色固体。
[0075]
s11.中间体17的制备
[0076][0077]
在室温下,将中间体15(1g,4.03mmol),丁二酸酐即化合物16(402mg,4.02 mmol)和diea(1g,8.04mmol)溶于10ml n,n-二甲基甲酰胺(dmf)中搅拌反应过夜。lc-ms显示反应完全。将反应液过滤后通过反相柱(甲酸fa,水:乙腈=100:0~85:15)分离纯化得中间体17(1g,收率:80%,3.2mmol)的白色固体。ms(esi)m/z(m+h)+313.0。
[0078]
s12.中间体18的制备
[0079][0080]
将中间体13(1g,1.2mmol)溶于20ml乙醇(etoh)中,加入二乙胺(5 ml)并在室温下继续搅拌反应5小时。tlc显示反应完全。将反应液浓缩,用 etoh带干两次。剩余物用乙酸乙酯(ea)溶解,通过少量的硅胶(200-300目) 过滤,用ea洗去杂质后,用加入二乙胺的甲醇洗涤硅胶。所得滤液旋干,用乙醇带干两次后得中间体18(650mg,收率:92.4%,1.11mmol)的黄色固体。 ms(esi)m/z(m+h)
+
587.3。1h nmr(400mhz,dmso)δ8.67(t,j=5.9hz,1h), 7.70(d,j=9.1hz,1h),7.44
–
7.32(m,2h),7.15(d,j=8.3hz,1h),6.99(t,j=5.5 hz,1h),6.64(dd,j=20.6,2.3hz,2h),6.47(dd,j=8.3,2.4hz,1h),4.27(d,j=5.7hz,2h),3.85(s,4h),3.73(s,4h),3.26
–
3.18(m,5h),2.72
–
2.67(m,4h),2.11
ꢀ–
2.03(m,4h),1.82
–
1.64(m,9h),1.46(d,j=19.2hz,7h)。
[0081]
s13.中间体19的制备
[0082][0083]
将中间体18(650mg,1.11mmol)、中间体17(346mg,1.11mmol)和diea (429mg,3.33mmol)溶于dmf(20ml)中,加入hatu(506mg,1.33mmol) 并在室温下继续搅拌反应1小时。lc-ms显示反应完全。加入水(50ml),用 ea(50ml
×
3)萃取,有机层用饱和食盐水洗涤(50ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经反相柱(fa,水:乙腈=100:0~50:50)分离纯化得中间体 19(527mg,收率:53.9%,0.599mmol)的橙色固体。ms(esi)m/z(m+h)
+
881.3。
[0084]
s14.mac-4c的制备
[0085][0086]
将中间体19(527mg,0.599mmol)溶于dcm(10ml)中,加入1ml三氟乙酸(tfa)并在室温下继续搅拌反应半小时。tlc显示反应完全。将反应液浓缩通过高效反相柱制备得mac-4c(200mg,收率:45.7%,0.273mmol)的黄色固体。ms(esi)m/z(m+h)
+
731.1。1h nmr(400mhz,dmso)δ8.67(t,j=5.8hz,1h),8.16(d,j=8.8hz,2h),7.68(d,j=9.0hz,1h),7.57(d,j=8.7hz, 2h),7.50(d,j=9.1hz,1h),7.37(s,1h),7.30(s,1h),7.25(dd,j=9.0,2.5hz,1h), 6.92(d,j=2.4hz,1h),6.03(s,2h),5.76(d,j=4.8hz,1h),5.03
–
4.95(m,1h), 5.00
–
4.77(t,1h),3.94(d,j=8.0hz,1h),3.56
–
3.48(m,1h),3.30
–
3.22(m,3h), 2.69(s,3h),2.18(dd,j=13.9,7.4hz,2h),2.14
–
2.03(m,8h),1.78(dt,j=14.4, 9.2hz,10h),1.48(s,2h)。
[0087]
2、一种mac-6c半抗原的制备方法,包括如下步骤:
[0088]
s1~s6同上mac-4c半抗原制备过程;
[0089]
s7.中间体21的制备
[0090][0091]
将boc保护的氨基己酸,即化合物20(5.66g,27.8mmol)、hatu(12.7g, 33.4mmol)和diea(10.8g,83.6mmol)溶于dmso(50ml)中并在室温下继续搅拌反应1小时。将上述液体缓慢滴加入正在搅拌的中间体9(10g,41.8mmol) 和diea(10.8g,83.6mmol)的dmso(100ml)溶液中,并在室温下继续搅拌反应半小时。lc-ms显示反应完全后。向反应液中直接加入fmoc-cl(21.63g, 83.6mmol)和diea(10.8g,83.6mmol),并继续搅拌反应1小时。lc-ms显示反应完全。加入水(300ml),用ea(100ml
×
5)萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经柱层析 (pe:ea=100:0~35:65)分离纯化得中间体21(8.2g,收率:48.9%,13.6mmol) 的淡黄色固体。ms(esi)m/z(m+h-100)
+
502.1。1h nmr(400mhz,dmso)δ 7.88(d,j=7.5hz,2h),7.70(s,2h),7.41(s,2h),7.34(dd,j=7.4,1.0hz,3h),7.10 (s,1h),6.74(t,j=5.3hz,1h),4.18(s,3h),4.03(q,j=7.1hz,1h),3.00
–
2.79(m, 2h),2.11(s,4h),1.90
–
1.70(m,8h),1.36(s,16h),1.18(t,j=7.1hz,4h)。
[0092]
s8.中间体22的制备
[0093][0094]
将中间体21(8.2g,13.6mmol)溶于4m的盐酸二氧六环溶液中(50ml) 中并在室温下继续搅拌反应1小时,脱除boc保护基,tlc显示反应完全后,将反应液浓缩得中间体22(8.12g)的淡黄色油状液体。ms(esi)m/z(m+h)
+ 502.3。1h nmr(400mhz,dmso)δ7.37(dt,j=32.7,7.4hz,1h),7.37(dt,j= 32.7,7.4hz,1h),4.18(s,1h),3.57(s,2h),2.75
–
2.71(m,
0h),2.69(s,1h),2.52
–ꢀ
2.48(m,1h),2.12(s,1h),2.01(t,j=7.3hz,0h),1.83(dd,j=36.3,23.0hz,1h), 1.50(dtd,j=22.5,15.0,7.5hz,1h),1.33
–
1.17(m,1h)。
[0095]
s9.中间体23的制备
[0096][0097]
将中间体6(1.8g,5.1mmol)、hatu(2.3g,6.1mmol)和diea(1.9g,15.3 mmol)溶于dcm(20ml)中并在室温下继续搅拌反应1小时。将上述液体加入正在搅拌的中间体22(2.6g,5.1mmol)的dcm(100ml)溶液中,并在室温下继续搅拌反应半小时。lc-ms显示反应完全后。加入水(100ml),用dcm (100ml
×
3)萃取,有机层用饱和食盐水洗涤(100ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经柱层析(pe:ea=100:0~0:100)分离纯化得中间体23(3.4 g,收率:80.4%,4.1mmol)的橙色固体。ms(esi)m/z(m+h)
+
837.2。1h nmr (400mhz,dmso)δ8.65(s,1h),7.88(d,j=7.4hz,2h),7.70(dd,j=8.2,4.0hz, 3h),7.40(dd,j=13.7,4.9hz,3h),7.32(td,j=7.4,1.0hz,3h),7.15(d,j=8.3hz, 1h),6.98(s,1h),6.63(dd,j=20.1,2.4hz,2h),6.46(dd,j=8.4,2.4hz,1h),5.76 (s,1h),4.27(d,j=5.6hz,2h),4.18(s,3h),3.84(s,3h),3.73(s,3h),3.28
–
3.21 (m,2h),2.69(d,j=1.4hz,4h),2.08(d,j=17.2hz,3h),1.79(d,j=20.5hz,7h), 1.58
–
1.41(m,7h),1.28(dd,j=19.1,12.1hz,7h)。
[0098]
s10.中间体24的制备
[0099][0100]
将中间体23(3.0g,3.6mmol)溶于etoh(20ml)中,加入二乙胺(5ml) 并在室温下继续搅拌反应5小时。tlc显示反应完全。将反应液浓缩,用etoh 带干两次。剩余物用ea溶解,通过少量的硅胶(200-300目)过滤,用ea洗去杂质后,用加入二乙胺的甲醇洗涤硅胶。所得滤液旋干,用乙醇带干两次后得中间体24(2.2g,收率:97.2%,3.5mmol)的黄色固体。ms(esi)m/z(m+h)
+ 615.3。1h nmr(400mhz,dmso)δ8.65(t,j=5.9hz,1h),7.70(d,j=9.1hz, 1h),7.39(dd,j=9.1,2.5hz,1h),7.25(s,1h),7.15(d,j=8.4hz,1h),6.98(t,j= 5.7hz,1h),6.63(dd,j=17.9,2.4hz,2h),6.47(dd,j=8.3,2.4hz,1h),5.32(t,j= 4.7hz,0h),4.39
–
4.19(m,3h),3.85(s,3h),3.73(s,3h),3.55
–
3.40(m,4h),2.69 (s,3h),1.74(d,j=16.2hz,6h),1.56
–
1.44(m,5h),0.85(t,j=6.7hz,1h)。
[0101]
s11.中间体26的制备
[0102][0103]
将己二酸单甲酯,即化合物25(2.19g,13.7mmol)、hatu(7.81g,20.6mmol) 和diea(8.85g,68.5mmol)溶于dmf(50ml)中并在室温下继续搅拌反应1 小时。将中间体15(2.9g,
13.7mmol)加入溶液中,并在室温下继续搅拌反应半小时。lc-ms显示反应完全。过滤后的液体用反相柱层析(fa,水:乙腈=100:0~90:10)分离纯化得中间体26(3.2g,收率:65.7%,9.0mmol)的白色固体。ms(esi)m/z(m+h)
+
355.2。
[0104]
s12.中间体27的制备
[0105][0106]
将中间体26(1.6g,4.5mmol)和lioh(0.4g,9mmol)溶于thf(20ml) 和水(10ml)中并在室温下继续搅拌反应1小时。lc-ms显示反应完全。反应液用浓盐酸调ph至中性,减压蒸馏除去thf后冻干得中间体27(1.8g)的白色固体,产物不用进一步纯化。ms(esi)m/z(m-h)-339.1。
[0107]
s13.中间体28的制备
[0108][0109]
将中间体24(2.2g,2.93mmol)、中间体27(1.1g,3.22mmol)和diea(1.13 g,8.79mmol)溶于dmf(20ml)中,加入hatu(1.34g,3.52mmol)并在室温下继续搅拌反应1小时。lcms显示反应完全。加入水(50ml),用ea(50 ml
×
3)萃取,有机层用饱和食盐水洗涤(50ml)后用无水硫酸钠干燥过滤后浓缩。剩余物经反相柱(fa,水:乙腈=100:0~50:50)分离纯化得28(1g,收率: 35.1%,1.0mmol)的橙色固体。ms(esi)m/z(m+h)
+
937.2。1h nmr(400mhz, dmso)δ8.66(s,1h),8.38(s,1h),8.15(d,j=8.7hz,2h),7.70(d,j=9.2hz,1h), 7.57(d,j=8.7hz,2h),7.50
–
7.34(m,2h),7.21(dd,j=43.5,15.5hz,4h),6.98(d, j=5.2hz,1h),6.63(dd,j=18.0,2.2hz,3h),6.56
–
6.43(m,1h),5.32(s,1h), 5.01(s,1h),4.84(s,1h),4.27(d,j=5.5hz,2h),3.97(s,1h),3.85(s,3h),3.73(s, 3h),3.51(s,2h),2.68(s,4h),2.11(s,2h),1.91
–
1.71(m,11h),1.63
–
1.39(m, 8h)。
[0110]
s14.mac-6c的制备
[0111][0112]
将中间体28(1g,1.06mmol)溶于dcm(20ml)中,加入tfa(2ml) 并在室温下继续搅拌反应5min。tlc显示反应完全。将反应液浓缩通过高效反相柱制备得mac-6c(350mg,收率:44.5%,0.44mmol)的黄色固体。ms(esi) m/z(m+h)
+
787.4。1h nmr(400mhz,dmso)δ8.65(t,j=5.8hz,1h),8.15(d,j =8.8hz,2h),7.68(d,j=9.0hz,1h),7.57(d,j=8.7hz,2h),7.44(d,j=9.2hz, 1h),7.29(s,1h),7.25(dd,j=9.0,2.5hz,1h),7.23(s,1h),6.91(d,j=2.4hz,1h), 6.02(s,2h),5.80(d,j=4.8hz,1h),5.01(dd,j=4.6,2.4hz,1h),4.82(dd,j=6.3, 4.7hz,1h),3.98(d,j=8.8hz,1h),3.54(dd,j=15.8,9.2hz,1h),3.30
–
3.22(m, 3h),2.68(s,3h),2.11(s,2h),2.07
–
1.93(m,6h),1.89(t,j=6.7hz,2h),1.86
–ꢀ
1.74(m,8h),1.59
–
1.43(m,6h),1.35
–
1.17(m,6h)。
[0113]
实施例2mac型多簇串联线性人工抗原的制备
[0114]
本实施例提供mac型多簇串联线性人工抗原的制备方法,主要包括免疫原和包被原的合成。免疫原与包被原的制备不同之处在于载体种类,所述的免疫原采用的载体牛乳铁蛋白(lf)、钥孔血蓝蛋白(klh);所述的包被原采用载体蛋白为卵清蛋白(ova)。免疫原的制备方法。本发明采用的合成免疫原/包被原的方法为重氮化法。具体步骤如下:
[0115]
称取mac-4c 22.09mg(mac-6c 23.79mg)溶于1ml水中,用0.1m hcl 调ph至2.0,加入10mg/ml nano2约350μl,4℃避光反应30min,记为a液。称取载体蛋白10mg溶于4ml cbs缓冲溶液(0.01m,ph 9.0)中,搅拌溶解制成b液。磁力搅拌下,吸取a液逐滴加入到b液中,4℃下磁力搅拌反应12h。将反应液于4℃下用pbs透析3天,每天换2次透析液。即可得到免疫原,冻存于-20℃冰箱中,备用。取实施例1制备的半抗原和实施例2制备的人工抗原分别进行紫外波长扫描(150~400nm)进行鉴定,结果如图1所示,相比于载体蛋白与半抗原,偶联后特征吸收峰及峰形均发生变化,结果说明抗原偶联成功。
[0116]
实施例3多簇串联线性人工抗原特异性多克隆血清制备
[0117]
动物免疫:用健康的6周龄的balb/c雌鼠作为实验动物,将实施例2制备的两种免疫原分别与等量的佐剂(第一次用完全弗氏佐剂,之后均用不完全弗氏佐剂)混合乳化,在小鼠颈背部和腹腔进行皮下注射,每次免疫剂量为0.5ml(其中含有0.5mg免疫原)。首次免疫用0.5ml完全弗氏佐剂与抗原乳化后用于免疫,4周后用0.5ml不完全弗氏佐剂与抗原乳化后加强免疫,之后每隔2周免疫一次,期间尾巴静脉取少量血进行抗体质量鉴定,待抗体稳定后,选择性能最好的小鼠收集血清,37℃温浴30min,室温离心20min,取上清分装于离心管中,于-20℃下保存使用。
[0118]
抗血清效果评价:以实施例2制备得到的包被抗原mac-4c-ova或 mac-6c-ova,取上述采集的的小鼠血清为检测抗体,采用间接竞争elisa法测定小鼠血清的抗血清效价与抑制率,综合考虑各抗血清的效价、抑制率,对其进行评价。具体操作步骤如下:
[0119]
1)包板:将包被抗原mac-4c-ova或mac-6c-ova用0.05m碳酸盐缓冲液(ph 9.6)稀释至1000ng/ml,按100μl/孔,4℃包被过夜;弃包被液,pbst 洗涤2次,每孔加入120μl封闭液(5%脱脂牛奶)37℃封闭3h;弃封闭液, 37℃烘干60min,用密封袋装于4℃待用,得到包好的酶标板。
[0120]
2)血清效价与抑制检测:将步骤1)包好的酶标板,效价列:每孔分别加入50μl pbs和50μl按梯度倍数稀释的血清;抑制列:每孔加入50μl稀释好的1000ng/ml药物(mac-4c或mac-6c)和50μl按梯度倍数稀释的血清,做2组平行。37℃孵育40min,用pbst洗五次,拍干孔内液体,加入1:5000 稀释的酶标二抗(羊抗鼠igg-hrp),37℃孵育30min,用pbst洗五次,拍干孔内液体,加入100μl tmb底物液,37℃避光显色10min;加入50μl终止液 (10%h2so4)终止反应;用酶标仪读取450nm处的吸光值。
[0121]
3)实验结果
[0122]
基于elisa法测定小鼠血清的抗血清效价与抑制率,结果如表1所示,效价在8~16k之间,对个1μg/ml浓度的mac-4c和mac-6c的抑制率为51~85%。
[0123]
表1小鼠抗血清效果
[0124][0125]
以上结果表明,本发明制备的mac型多簇串联线性半抗原及其人工抗原具有良好的免疫原性,可以用于后续制备单克隆抗体、单链抗体和纳米抗体。
[0126]
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1