注射成型零件的制作方法
注射成型零件
1.本发明涉及一种注射成型零件,所述注射成型零件包含含有聚亚芳基硫醚和玻璃纤维的组合物。特别地,本发明涉及包括注射成型零件的燃料电池应用。本发明还涉及一种用于制备包含聚亚芳基硫醚和玻璃纤维的组合物的方法,以及涉及一种用于制备包含所述组合物的注射成型零件的方法。
2.燃料电池,特别是质子交换膜燃料电池(proton exchange membrane fuel cell,pemfc),是一种将氢燃料与氧组合以产生电、热和水的电化学装置。多个燃料电池被称为燃料堆。燃料堆可以包括数百个单独的燃料电池以及由各种材料(例如聚合物组合物和/或金属)制成的各种部件。每个单独的燃料电池包含双极板、气体耗散层和具有铂催化剂层的质子交换膜的夹层结构。铂催化剂氧化氢分子,让它选择性地将氢离子从阳极传递到阴极,并迫使电子作为电流行进通过外部设备到达阴极。在给定燃料电池中化学反应的性质的情况下,从用于制造燃料电池堆的部件的材料中浸出的离子必须被最小化并理想地被阻止。从燃料电池堆中使用的部件浸出的杂质和离子可能使催化剂中毒并且可能堵塞膜,这可能显著降低燃料电池堆的效率并影响其使用寿命。
3.作为注射成型零件的含有聚亚芳基硫醚和玻璃纤维的用于燃料电池的任何部件,应该表现出低离子浸出,以便维持燃料电池堆的效率。此外,特别是当与水接触时,这些注射成型零件应该表现出足够的水解稳定性,特别是在高温下足够的水解稳定性。燃料电池工作温度通常介于50℃与80℃之间,其中峰值温度为约110℃,并且这使得注射成型零件成为必要,所述注射成型零件组合了低离子浸出与足够的水解稳定性,例如足够的断裂伸长率和拉伸强度,即使在高温下暴露于水中达较长的时间段之后也如此。
4.本发明的目的是提供注射成型零件,所述注射成型零件显示出较低的离子浸出和足够的机械保持,特别是在水解环境下,例如显示出足够的断裂伸长率和/或拉伸强度,特别是在高温下暴露于水或水/乙二醇之后。令人惊讶的是,这已经用包含组合物的注射成型零件实现,所述组合物包含:
5.a.聚亚芳基硫醚(polyarylene sulfide,pas),量介于50重量%与90重量%之间;
6.b.玻璃纤维,量介于10重量%与50重量%之间;
7.其中所述组合物具有如通过电感耦合等离子体原子发射光谱法(inductively coupled plasma atomic emission spectroscopy,icp-aes)测量的至多3500ppm的钠含量,并且其中所述组合物具有如通过x射线荧光(x-ray fluorescence,xrf)测量的至多100ppm的碘含量,并且其中重量百分比和ppm是相对于所述组合物的总重量而言的。
8.us2018265701涉及一种树脂组合物,所述树脂组合物包含具有降低的氯含量和降低的钠含量的聚亚芳基硫醚树脂和填料。然而,所述组合物具有高碘含量,因为聚亚芳基硫醚是采用苯的碘化并进一步与元素硫反应以形成聚亚苯基硫醚而制备的。这具有的缺点是在这种聚合方法中,碘回收在这种方法中几乎不能完全实现,从而导致聚亚芳基硫醚的高成本。此外,嵌在聚合物链中和/或在端基处的碘部分在聚亚芳基硫醚的每次热处理中可进一步有活性。
9.注射成型零件本身是已知的,并且通过本领域技术人员已知的注射成型工艺获
得。注射成型包括以下步骤:将包含pas的组合物加热至高于pas的熔融温度以获得熔体,用所述熔体填充模具,随后冷却所述模具和组合物,使得所述组合物固化成注射成型零件。
10.根据本发明的注射成型零件包含组合物,所述组合物含有介于50重量%与90重量%之间的聚亚芳基硫醚(pas),其中所述重量百分比是相对于组合物的总重量而言的。优选地,pas以介于55重量%与85重量%之间,更优选地介于60重量%与80重量%之间,最优选地介于60重量%与70重量%之间的量存在。在一个优选的实施方式中,pas是聚(对亚苯基)硫醚(poly(p-phenylene)sulfide,pps),因为pas具有pps已有的优点。
11.根据本发明的注射成型零件包含组合物,其中,相对于所述组合物的总重量,所述组合物的钠含量为至多3500ppm,优选至多3000ppm,甚至更优选至多2500ppm,并且最优选至多2000ppm。钠含量可以用电感耦合等离子体原子发射光谱法(icp-aes)来测量,如下所述。所述组合物的钠含量可以低至20ppm。
12.优选地,根据本发明的注射成型零件包含组合物,所述组合物表现出至少230℃,更优选至少235℃;最优选至少240℃的结晶温度(tc),所述结晶温度测量如下:根据iso 11357-1/3(2009)的方法,通过dsc,以10℃/min的扫描速率将组合物加热至320℃,并将所述组合物在氮气下于320℃保持3分钟,随后以相同的扫描速率冷却所述组合物以记录第一冷却循环中的冷却结晶温度。这具有提高注射成型零件的水解稳定性的优点。
13.优选地,根据本发明的注射成型零件包含pas,更优选pps,钠含量为至多500ppm,更优选至多400ppm,最优选至多300ppm,甚至更优选至多250ppm的量,其中ppm是分别相对于pas或pps的总重量而言。钠含量可以低至5ppm。
14.根据本发明的注射成型零件包含组合物,其中pas的主要结构为-(ar-s)-(ar是亚芳基)作为重复单元。亚芳基的示例是对亚苯基、间亚苯基、经取代的亚苯基、p,p'-二亚苯基醚基、p,p'-二亚苯基羰基、和萘基。pas可以通过本领域技术人员已知的方法聚合。特别优选的生产方法包括在有机极性溶剂中聚合硫源和二卤代芳族化合物以生产聚亚芳基硫醚的聚合步骤。pps的所述生产方法由美国专利第3,919,177号公开。所述生产方法不在pas中生成任何链结合的和/或任何游离的碘。所得pas,例如pps,不含碘,或者如果存在碘的话,则碘含量低于10ppm,优选低于5ppm。如上所述的pas的低钠含量可以通过用酸洗来实现。酸洗是一种本身已知的工序。在pas聚合后,所述pas优选通过用酸洗涤、用热水洗涤、或用有机溶剂洗涤、或它们的组合来处理,以将pas的端基从-sna改变为-sh。优选地,洗涤溶液的ph值介于2与7之间,合适的洗涤溶液可以是乙酸(ch3cooh)、磷酸(h3po4)和草酸(c2h2o4)或其他有机酸,更优选地,使用乙酸。
15.此外,优选地,pas,优选pps的结晶温度(tc)为至少230℃,更优选至少235℃;最优选至少240℃,所述结晶温度测量如下:根据iso 11357-1/3(2009)的方法,通过dsc,以10℃/min的扫描速率将组合物加热至320℃,并将所述组合物在氮气下于320℃保持3分钟,随后以相同的扫描速率冷却所述组合物以记录第一冷却循环中的冷却结晶温度而测量的,因为这提高了注射成型零件的水解稳定性。
16.优选地,pas的重均分子量(mw)在10000-100000g/mol的范围内,更优选地在20000-80000g/mol的范围内,甚至更优选地在30000-80000g/mol的范围内;最优选地在30000-70000g/mol的范围内。
17.优选地,适用于本技术的聚亚芳基硫醚的pdi(重均分子量/数均分子量;mw/mn)小
苯基-γ-氨基丙基三乙氧基硅烷和n-苯基-γ-氨基丙基三甲氧基硅烷,优选地所述氨基烷氧基硅烷是γ-氨基丙基三乙氧基硅烷和/或γ-氨基丙基三甲氧基硅烷。
24.注射成型零件包含组合物,所述组合物包含:
25.a.聚亚芳基硫醚(polyarylene sulfide,pas),量介于50重量%与90重量%之间;
26.b.玻璃纤维,量介于10重量%与50重量%之间;
27.其中重量百分比是相对于组合物的总重量而言;并且其中所述组合物的钠含量为至多3500ppm,如通过icp-aes测量的。所述组合物的钠含量为优选至多3000ppm,更优选至多2500ppm,甚至更优选至多2000ppm。所述组合物的钠含量可以低至20ppm。
28.pas中、复合组合物或注射成型零件中的钠含量是通过以下方法用icp-aes测量的:
29.由于钠在燃烧期间保持稳定,因此通过灰渣法(ash residue method)制备样本。
30.步骤1:在陶瓷坩埚中准确地称量约5克样本,并使用本生灯(bunsen burner)缓慢燃烧。然后将经燃烧的残渣放入600℃的马弗炉中3小时,以确保完全焚烧。再次称量坩埚以量化灰分含量,因为灰分百分比用于重新计算原始样本中的实际钠浓度。
31.步骤2:使用白金实验室器皿,于1250℃将约1克的灰渣与5克偏硼酸锂一起熔融;称量的量均是准确称量的。
32.步骤3:随后使用摇床将精确称量的约1克熔融材料溶解在10ml h2so4和10ml h2o中16h。
33.步骤4:将所溶解的溶液进一步用h2o稀释至100ml。
34.步骤5:使用来自thermo scientific的icap6500光谱仪,借助于icp-aes分析所获得的溶液。根据校准线执行测量,所述校准线是使用来自alfa aesar的经认证参考溶液制备的。
35.注射成型零件包含组合物,所述组合物的碘含量为至多100ppm,如通过x射线荧光(x-ray fluorescence,xrf)测量的。优选地,碘含量为至多80ppm,更优选地至多70ppm,最优选地至多50ppm。所述组合物的碘含量可能非常低,并且因此低于xrf方法的检测限,所述检测限通常为约20ppm。
36.碘含量是通过x射线荧光(xrf)测量的。因为碘由于其不稳定性而无法通过样本燃烧法进行测量,所以碘是通过xrf分析直接在原样的原始聚合物中或在组合物中分析的。以完全覆盖量杯底部(直径40mm,4mm厚)的方式冲压零件,例如平坦的饰板,例如拉伸条。然后使用来自panalytical的配备有rh x射线管的axios max先进wdxrf光谱仪,借助于xrf分析饰板。对参考样本进行共分析,以确认碘信号的正确位置。
37.在一个优选的实施方式中,注射成型零件包含组合物,所述组合物还包含量为相对于所述组合物的总重量,量介于0.1重量%与1.0重量%之间的偶联剂。令人惊讶的是,这导致组合物的水解稳定性进一步提高。
38.偶联剂本身是已知的,并且具有通式(i):
39.(x-(ch2)
x
)
y-si-(o-c
nh(2n+1)
)
(4-y)式(i)
40.其中取代基定义如下:
41.x是nh2。
42.x是1至10的整数,优选2或3;
43.y是0至3的整数,优选0或3;
44.n是1至3的整数,优选1或2。
45.合适的偶联剂包括例如γ-氨基丙基三乙氧基硅烷、γ-氨基丙基三甲氧基硅烷、γ-氨基丙基甲基二乙氧基硅烷、γ-氨基丙基甲基二甲氧基硅烷、n-β(氨基乙基)-γ-氨基丙基三乙氧基硅烷、n-β(氨基乙基)-γ-氨基丙基-三甲氧基硅烷、n-β(氨基乙基)-γ-氨基丙基甲基二甲氧基硅烷、n-苯基-γ-氨基丙基三乙氧基硅烷和n-苯基-γ-氨基丙基三甲氧基硅烷,优选地所述氨基烷氧基硅烷是γ-氨基丙基三乙氧基硅烷和/或γ-氨基丙基三甲氧基硅烷。可以在制备组合物时投加偶联剂,这通常通过在挤出机中混合来完成。优选地,将偶联剂与玻璃纤维一起在侧进料处投加。其他合适的偶联剂是例如脲基丙基三甲氧基硅烷或脲基丙基三乙氧基硅烷,如在us2015/0166731a1中所公开的。令人惊讶的是,偶联剂的添加导致组合物表现出进一步的水解稳定性。
46.本发明还涉及一种燃料电池,所述燃料电池包括根据上文所公开的实施方式中的任一实施方式的注射成型零件。这些注射成型部件包括例如但不限于介质分配板、歧管、绝缘板、介质连接器、气流控制阀、气流断路器、氢气注入器、氢气供应阀、氢气调节阀、压力控制阀、氢气循环泵、加湿器、恒温器、电控冷却阀、电控冷却剂泵。
47.本发明的一个实施方式涉及如上所公开的注射成型零件,其中,所述组合物针对厚度为4mm的注射成型拉伸条上表现出至少170mpa,优选地至少175mpa,更优选地至少180mpa的拉伸强度,所述拉伸强度测量如下:如根据iso 527-1a,以5mm/min,于23℃下,暴露于135℃温度的水乙二醇(w/g)混合物(50%/50%体积%/体积%)1000小时期间之后。在另一个实施方式中,本发明涉及如上所公开的注射成型零件,其中,所述组合物针对厚度为4mm的注射成型拉伸条表现出至少1.5%,优选地至少1.6%,更优选地至少1.7%的断裂伸长率,所述断裂伸长率测量如上:根据iso 527-1a,以5mm/min,于23℃下,暴露于135℃温度的水乙二醇(w/g)混合物(50%/50%体积%/体积%)1000小时期间之后。在一个优选的实施方式中,注射成型零件表现出如上所公开的拉伸强度和断裂伸长率的组合。所有单个范围都是明确可组合的。令人惊讶的是,所述注射成型零件组合了在高温下暴露于水/乙二醇之后的足够拉伸强度和断裂伸长率,与此同时减少了离子浸出。这允许特别是其中注射成型部件可能与含水流体接触的应用。
48.在一个优选的实施方式中,本发明涉及如上所公开的注射成型零件,其中,所述组合物针对厚度为4mm的注射成型拉伸条表现出至少160mpa,优选地至少165mpa,更优选地至少170mpa的拉伸强度,所述拉伸强度测量如下:如根据iso 527-1a,以5mm/min,于23℃下,暴露于高压釜中的110℃温度的水蒸气1000小时期间之后。在另一个实施方式中,本发明涉及如上所公开的注射成型零件,其中,所述组合物针对厚度为4mm的注射成型拉伸条表现出至少1.2%,优选地至少1.3%,更优选地至少1.4%,甚至更优选地至少1.5%,最优选地至少1.6%的断裂伸长率,所述断裂伸长率测量如下:根据iso 527-1a,以5mm/min,于23℃下,在暴露于110℃温度的水1000小时期间之后。在一个优选的实施方式中,注射成型零件表现出如上所公开的拉伸强度和断裂伸长率的组合。所有单个范围都是明确可组合的。在燃料电池应用中,注射成型零件可以在高温下与水接触,并且令人惊讶的是,根据本发明的注射成型零件表现出足够的断裂伸长率和拉伸强度,与此同时减少了离子的浸出。
49.本发明进一步涉及一种用于制备包含聚亚芳基硫醚和玻璃纤维的组合物的方法,
其中所述组合物具有如通过电感耦合等离子体原子发射光谱法(icp-aes)测量的为至多3500ppm的钠含量,并且其中所述组合物具有如通过x射线荧光(xrf)测量的为至多100ppm的碘含量,并且其中重量百分比和ppm是相对于所述组合物的总重量而言的,所述方法包括以下步骤:例如用挤出机将pas加热至高于其熔融温度的温度,随后加入玻璃纤维以获得混合物,在此之后将混合物冷却并可适当造粒。优选地,如果组合物进一步包含偶联剂,则将所述偶联剂与玻璃纤维一起在侧进料处投加到pas中,更优选地所述偶联剂是氨基烷氧基硅烷、γ-氨基丙基三乙氧基硅烷和/或γ-氨基丙基三甲氧基硅烷。如上所公开的所有优选范围也适用于用于制备组合物的方法。
50.本发明还涉及一种组合物,所述组合物包含:
51.a.聚亚芳基硫醚(polyarylene sulfide,pas),量介于50重量%与90重量%之间;
52.b.玻璃纤维,量介于10重量%与50重量%之间;
53.其中所述组合物具有如通过电感耦合等离子体原子发射光谱法(inductively coupled plasma atomic emission spectroscopy,icp-aes)测量的为至多3500ppm的钠含量,并且其中所述组合物具有如通过x射线荧光(x-ray fluorescence,xrf)测量的为至多100ppm的碘含量,并且其中重量百分比和ppm是相对于所述组合物的总重量而言的。如上所公开的包含所述组合物的注射成型零件的所有优选范围也适用于涉及所述组合物的本发明。
实施例:
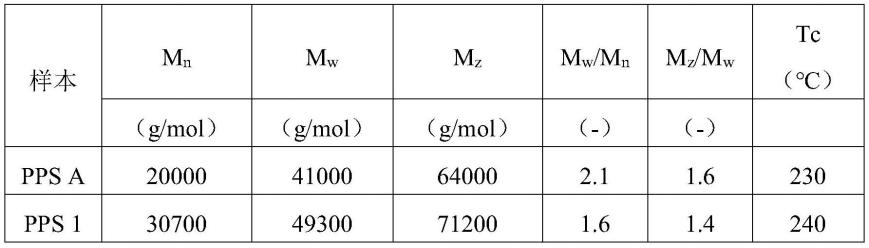
54.所使用的材料:
55.玻璃纤维:
56.neg ecs03t-747h/r,可从neg获得,其中关于玻璃纤维的钠含量为4500ppm。碘含量低于检测限。
57.ds8800-11p 4mm,可从3b获得,其中关于玻璃纤维的钠含量为400ppm。碘含量低于检测限。
58.pps:
59.pps a和pps 1是根据如在美国专利第3,919,177号中描述的方法生产的。在此方法中,对二氯苯与nahs在n-甲基-2-吡咯烷酮溶剂中在约250℃的高温下反应,直到达到期望的mw。pps 1在聚合后进一步经历用水于80℃洗涤的步骤,以通过将-sna端基部分转化为-sh来降低-sna端基的量,这导致低钠含量。
60.pps a和pps 1的分子特征、结晶温度、钠和碘含量是根据如以上说明书中所述的方法测定的。
61.结果如下所示。
[0062][0063]
pps a的钠含量为1500ppm,相对于pps的总重量为0.15重量%。
[0064]
pps 1的钠含量为400ppm。
[0065]
pps a和pps 1的碘含量低于检测限。
[0066]
比较材料b:a504x90(c),从toray获得。含有钠含量为1700ppm的pps,基于pps和40重量%的玻璃纤维的总重量。比较材料b的碘含量低于检测限。
[0067]
比较材料c:1140l4,可从celanese获得。这种组合物含有40重量%的玻璃纤维,并且钠含量为基于组合物的总重量,0.47重量%(4700ppm)。比较材料c的碘含量低于检测限。
[0068]
表1组合物
[0069][0070]
除了从toray获得的比较b和从celanese获得的比较c之外,通过混合如表1中所呈现的成分来制备组合物。
[0071]
将pps和偶联剂的混合物与玻璃纤维组合,以避免玻璃纤维断裂,并使用双螺杆挤出机在约315℃至约420℃的温度下熔融配混。将熔融组合物挤出成线料,并在切成粒料之前穿过水浴。将所得粒料于140℃干燥至少4小时,然后通过在315℃至345℃的熔融温度和135℃至150℃的模腔表面温度下注射成型,模制成测试例如拉伸强度测试、拉伸模量测试、
拉伸应变测试的测试制品。
[0072]
表2中的所有拉伸测试都是根据标准测试方法iso 527-2进行的。对测试制品进行拉伸测试以获得初始特性值(t0小时值),并且数据显示在表2-1至表2-6中。使测试制品经受水乙二醇(w/g)混合物(50%/50%体积%/体积%)。如表2-1至表2-3所示,通过在不同的时间段(例如,1周、2周和6周)内将测试制品(例如,模制的测试试样)完全浸入使用蒸汽加热至135
±
2℃的封闭不锈钢压力容器内的w/g中以产生老化的测试制品,来进行测试制品的w/g老化。然后回收老化的测试制品并进行拉伸测试以获得最终特性值,并且数据显示在表2-1至表2-3中(拉伸特性,于135℃老化,于23℃测试)。表格中的“n.m.”代表未测出。
[0073]
表2-1在23℃的拉伸测试iso 527-1a 5mm/min-拉伸模量
[0074][0075]
表2-2在23℃的拉伸测试iso 527-1a 5mm/min-拉伸强度
[0076][0077]
表2-3在23℃的拉伸测试iso 527-1a 5mm/min-断裂伸长率(elongation at break,eab)
[0078][0079]
于110℃的高压釜老化
[0080]
对测试制品进行拉伸测试以获得初始特性值,在表中称为t0,并且数据显示在表2-4至表2-6中。如表2-4至表2-6所示,使测试制品经受高压釜中110℃的水蒸气达不同的时间段,即在500小时和1000小时后,以产生老化的测试制品。然后回收老化的测试制品并进行拉伸测试以获得最终特性值,并且数据在表2-4至表2-6中显示,展示了于110℃老化和于23℃测量的拉伸特性。
[0081]
表2-4在23℃测量的拉伸测试iso 527-1a 5mm/min-拉伸模量
[0082][0083]
表2-5在23℃测量的拉伸测试iso 527-1a 5mm/min-拉伸强度(tensile strength)
[0084][0085]
表2-6在23℃测量的拉伸测试iso 527-1a 5mm/min-eab
[0086][0087]
表2-1至表2-3清楚地表明,各种样本的弹性模量是相似的。实施例1的拉伸强度和eab最高,并且在长时间暴露于w/g后仍保持较高。比较b显示出拉伸强度和eab的急剧下降,在1008小时后不再测量到所述拉伸强度和eab。
[0088]
表2-4至表2-6显示了不同样本的弹性模量是相似的。同样在这里,实施例1的拉伸强度和eab最高,并且在长时间暴露于水蒸气后也保持较高。比较a和比较c显示出拉伸强度和eab的急剧下降,与实施例1相反,实施例1的拉伸强度和eab甚至在1000小时后仍然足够。
[0089]
浸出实验
[0090]
样本信息
[0091]
三种组合物作为拉伸条用于浸出实验,即比较a、比较b和实施例1,如表1中所述。使用1/2拉伸条的测试试样。测试试样具有以下特性:4.0mm厚,总表面积32cm2。根据iso527-1a测试试样。
[0092]
浸出温育方案:
[0093]
1)将试样放入100ml超纯水中(=32mm2/ml);
[0094]
2)在密闭的teflon
tm
feb瓶中于90℃进行烘箱老化;
[0095]
3)于90℃温育6周,对100ml进行icp-aes测量,还包括在teflon瓶中用blanco温育的100ml参考样本。
[0096]
用于浸出结果的icp-aes分析装置
[0097]
取样约15ml液体用于icp-aes筛选,在测量前用0.5ml hno3酸化样本。使用经认证的参考标准执行定量多元筛选。使用来自thermo scientific的icap6500 icp-aes执行测量。表3中呈现了浸出元素si、ca、al、k、和na。在不同的pps样本之间观察到了浸出行为的显著差异。
[0098]
表3浸出结果
[0099][0100]
比较b清楚地显示了所有报道的元素中最差的浸出性能,紧随其后的是比较a。实施例1清楚地表明了所有报道元素中最低的浸出含量。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1