用于改进衣物洗涤剂去污力的两性改性低聚丙烯亚胺乙氧基化物的制作方法
用于改进衣物洗涤剂去污力的两性改性低聚丙烯亚胺乙氧基化物
1.本发明涉及某些两性改性的乙氧基化低聚胺及其制造和用途。
2.在下文中,低聚/聚丙烯亚胺也缩写为“ppi”,低聚/聚乙烯亚胺也缩写为“pei”。乙氧基化低聚/聚丙烯亚胺被称为“eppi”(或“eppi”),乙氧基化低聚/聚乙烯亚胺被称为“epei”(或“epei”)。
3.此外,在下文中,环氧乙烷有时被称为“eo”,环氧丙烷有时被称为“po”。
4.烷氧基化低聚和聚烯亚胺,尤其是烷氧基化低聚和聚乙烯亚胺众所周知为用于各种应用的添加剂,如衣物洗涤剂(例如ep3301154、ep3167034和ep112593),用于水泥基粘合剂体系的添加剂(wo2016187085)或硬表面清洁剂(ep2961819)。在用于洗衣应用的情况下,已经提到对初次清洗和二次清洗性能的益处,但尚未公开对流变学的任何具体影响。
5.此外,存在关于烷氧基化低聚和聚烯亚胺,尤其是关于烷氧基化低聚和聚乙烯亚胺的一些报道,描述了对制剂粘度的影响。us20180216037和us20190024026都描述了与环氧乙烷和环氧丙烷反应的聚乙烯亚胺(例如pei-(eo)
10
(po)5)作为含阴离子表面活性剂的制剂的粘度改进剂。pei烷氧基化物用于降低表面活性剂制剂的粘度。相比之下,尚未公开对清洁性能的益处。
6.此外,wo2020/030469描述了具有高分子量聚乙烯亚胺核的乙氧基化聚乙烯亚胺,其通过使用特定工艺(强欠羟乙基化(strong under-hydroxyethylation))制备以获得具有良好性能状况并同时表现出降低的对洗涤剂制剂粘度的负面影响的用于洗衣应用的添加剂。
7.相比之下,改性低聚和聚烯亚胺烷氧基化物较不常见。
8.ep2662436描述了阳离子烷氧基化聚乙烯亚胺与漂白促进剂组合用于自动洗碗应用。在wo2004020563中描述了用于洗涤剂组合物的疏水改性和两性改性的聚烯亚胺。
9.例如在ep2961821中描述了含有烷氧基化和任选进一步阳离子改性或两性改性的每分子带有至少6个n原子的聚烯亚胺的洗衣组合物。更具体地,公开了改性和未改性的每分子具有至少6个n原子的低聚和聚丙烯亚胺,其带来去污益处并有助于改善白度。尚未提到对洗涤剂流变学的任何影响。
10.此外,ep3039109和ep3039057描述了具有低熔点的主要乙氧基化和任选进一步两性改性的低聚和聚烯亚胺。由于将至少1摩尔的c3至c5环氧烷并入主要基于环氧乙烷的聚烷氧基化物链中,可获得具有低熔点的聚合物,这有利于它们的制备工艺。尚未提到与仅基于环氧乙烷并因此具有较高熔点的相同聚合物相比在应用性能方面的任何益处。
11.此外,ep1198492描述了用作衣物洗涤剂的添加剂的具有改进的热稳定性的两性离子(即两性改性的)烷氧基化聚胺。一般而言,通过独立的季铵化和硫酸化步骤的组合获得该材料的两性改性,其中硫酸化用氯磺酸之类的硫酸化剂进行。所述两性离子聚胺具有净阴离子电荷,即阳离子基团数超过阴离子基团数至少20%,优选至少50%。尚未提到对清洁性能的改进的益处和对洗涤剂粘度的积极影响。
12.制造烷氧基化和进一步两性改性的低聚和聚烯亚胺的方法原则上是现有技术中
已知的。
13.如上文提到,在ep1198492中描述了烷氧基化的两性离子聚胺的制造方法,其中该烷氧基化物已用改性剂(1)季铵化和使用不同的改性剂(2)(例如氯磺酸)硫酸化。所得结构具有净阴离子电荷。
14.wo2001029112描述了一种使用三氧化硫作为硫酸化剂对烷氧基化的季铵化聚胺进行硫酸化以实现高硫酸化程度的方法。所述结构也具有净阴离子电荷。
15.在ep 1309546中描述了一种由叔氨基醇开始的基于烷氧基化、季铵化和转硫酸化(transsulfation)的组合的方法。该方法包括叔胺与硫酸化前体(例如硫酸二烷基酯)的反应以形成包含季铵化胺和硫酸化物类的混合物。然后在酸性条件下将羟基物类硫酸化。更具体地,已经公开了通过使用辅助胺制备烷氧基化、季铵化和完全硫酸化的低聚和聚烯亚胺。所得聚合物不再含有任何游离羟基,即所有基团已转化成硫酸基团。因此,所述聚合物的净电荷是阴离子的。
16.在wo200424858中描述了基于不使用辅助胺的烷氧基化、季铵化和转硫酸化的组合的另一方法。该方法包括烷氧基化低聚或聚胺的主链中的叔胺与硫酸化前体(例如硫酸二烷基酯)的反应以形成包含季铵化聚胺烷氧基化物和硫酸化物类的混合物。然后在酸性条件下将羟基物类硫酸化。根据转硫酸化过程的转化程度,可获得表现出中性或弱净阳离子电荷的聚合物。在任何情况下,聚环氧烷嵌段除阴离子硫酸基团外还含有游离末端羟基。在上述专利ep2961821、ep3039109和ep3039057中也描述了用于特定聚胺主链和特定聚环氧烷侧链的相同方法。
17.尽管在现有技术中已经描述了在洗衣应用中表现出良好清洁性能并在一些情况下也没有对洗涤剂制剂的流变学造成巨大负面影响的几种不同的未改性和改性的低聚和聚烯亚胺烷氧基化物和它们的制造方法,但仍然需要更好的材料,尤其是在更低剂量下具有更好的性能(聚合物的重量效率改进)。此外,在洗衣应用中需要具有良好或甚至改进的清洁性能的聚合物和对洗衣制剂的粘度的低或甚至无(负面)影响。此外,还需要简单的制备方法,以避免使用多于一种改性剂进行季铵化和硫酸化和/或避免使用辅助材料。
18.出乎意料地,在本发明中已经发现,特定的两性改性的低聚丙烯亚胺乙氧基化物可有助于显著改进衣物洗涤剂的去污力,同时对制剂粘度的负面影响最小化。本发明的材料与现有技术聚合物相比的改进性能在极低添加量下尤其明显,这是用于上开门洗衣机的重要先决条件。它们的更高重量效率有利于它们以高浓缩产品形式,例如在单个单剂量中使用。本发明的两性改性的低聚丙烯亚胺乙氧基化物可容易地由各自的低聚胺开始通过乙氧基化、季铵化和转硫酸化的组合获得,而季铵化和转硫酸化使用硫酸二烷基酯作为单一改性剂进行,即不需要使用多于一种改性剂和/或不需要使用辅助材料。本发明的两性改性的低聚丙烯亚胺乙氧基化物具有中性或弱净阳离子电荷,这取决于在转硫酸化步骤过程中的转化程度。
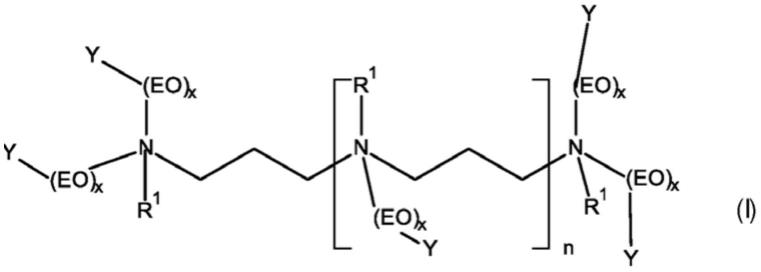
19.因此,本发明的一个方面是通式(i)的两性改性的低聚丙烯亚胺乙氧基化物(a)
[0020][0021]
其中
[0022]
r1相同或不同并选自c
1-c
4-烷基、h和自由电子对,其中所有r1的至少50%,优选至少80%,更优选至少90%是c
1-c
4-烷基,
[0023]
eo是指-ch
2-ch
2-o,
[0024]
y相同或不同并选自so
3-和h,其中所有y的至少30%,优选至少50%是so
3-,
[0025]
x相同或不同并选自5至50,优选10至40,且n选自1、2和3。
[0026]
本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)是内部两性离子。季铵化氮原子的抗衡离子是so
3-离子或烷基硫酸根离子(c1至c4单烷基硫酸根)。
[0027]
在任选中和和任选用水稀释后,该混合物中存在进一步的阴离子和阳离子,但该聚合物本身仍可被视为内部两性离子。
[0028]
在一个优选实施方案中,所有r1的93%至97%是c
1-c
4-烷基。
[0029]
在本发明的通式(i)的两性改性的低聚丙烯亚胺乙氧基化物(a)的一个优选实施方案中,n是2或3和/或所有r1的至少90%是甲基。
[0030]
在本发明的通式(i)的两性改性的低聚丙烯亚胺乙氧基化物(a)的进一步优选的实施方案中,n是2和/或所有r1的至少90%是甲基和/或x在15至30的范围内。
[0031]
在本发明的聚合物的进一步优选的实施方案中,n=1、2或3,r1的至少80%是c
1-c
4-烷基且r1=c
1-c
4-烷基与y=so
3-的比率为平均1.0:1.0至1.0:0.8。
[0032]
本发明的进一步方面包括:
[0033]
化合物混合物,其包含至少一种根据其中n=2的式(i)的两性改性的低聚丙烯亚胺乙氧基化物(a)和至少一种根据式(ii)的异构化合物
[0034][0035]
其中
[0036]
r1相同或不同并选自c
1-c
4-烷基、h和自由电子对,其中所有r1的至少50%,优选至少80%,更优选至少90%是c
1-c
4-烷基,
[0037]
eo是指-ch
2-ch
2-o,
[0038]
y相同或不同并选自so
3-和h,其中所有y的至少30%,优选至少50%是so
3-,
[0039]
x相同或不同并选自5至50。
[0040]
如上所述的优选的化合物混合物包含总摩尔比为10:1或更高的根据式(i)的两性改性的低聚丙烯亚胺乙氧基化物(a)和根据式(ii)的化合物。
[0041]
在本发明的一个优选实施方案中,本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)在另外包含碱金属和/或胺的硫酸盐的混合物中获得。
[0042]
在本发明的化合物混合物的进一步优选的实施方案中,该混合物另外包含胺的硫酸盐,优选链烷醇胺的硫酸盐。
[0043]
在本发明的另一优选实施方案中,本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)和含有其的混合物以任何比率与水混合。优选地,该混合物含有1至80重量%水,更优选1至60重量%水,再更优选5至50重量%水,最优选10至40重量%水。
[0044]
也可向本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)和含有其的混合物的水溶液中加入至少一种抗微生物剂。优选地,使用2-苯氧基乙醇(cas-no.122-99-6,例如可获自basf的pe)或4,4
’‑
二氯-2-羟基二苯醚(cas:3380-30-1)及其组合。
[0045]
4,4
’‑
二氯-2-羟基二苯醚可作为溶液使用,例如30重量%的4,4
’‑
二氯-2-羟基二苯醚在1,2-丙二醇中的溶液,例如可获自basf的hp 100。
[0046]
制造本发明的化合物的方法
[0047]
本发明的进一步方面还是一种制造本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)的方法,所述方法包括如下步骤:
[0048]
(a)提供选自氨、1,3-丙二胺、双-(3,3
’‑
氨基丙基)胺和双-(3,3
’‑
氨基丙基)-1,3-丙二胺或其混合物的胺,
[0049]
(b)任选地,所述胺用丙烯腈以100:1至1:2.5,优选10:1至1:2.5,更优选3:1至1:2.1的比率进行氰乙基化,随后氢化,以获得具有2、3和4个重复单元的低聚丙烯亚胺,
[0050]
(c)任选地,来自步骤(b)的低聚丙烯亚胺的纯化,
[0051]
(d)来自步骤a、b或c的所述胺和/或低聚丙烯亚胺的乙氧基化,和
[0052]
(e)用硫酸二-c
1-c
4-烷基酯至少部分季铵化和转硫酸化。
[0053]
在本发明的方法的一个优选实施方案中,进行纯化步骤(c)以获得纯度为至少80重量%,优选至少90重量%的具有2、3和4个重复单元的低聚丙烯亚胺或其混合物。
[0054]
在上述本发明的方法的一个优选实施方案中,步骤(e)中的季铵化用硫酸二甲酯进行。
[0055]
在上述本发明的方法的进一步优选的实施方案中,步骤(e)中的转硫酸化用硫酸作为催化剂进行。
[0056]
在本发明的方法的进一步优选的实施方案中,步骤(e)中的转硫酸化定量进行(》=80%)并获得弱阳离子或净中性聚合物。
[0057]
在上述本发明的方法的进一步优选的实施方案中,该方法另外包括用选自碱金属
氢氧化物和胺的碱中和硫酸的后续步骤。优选地,可使用选自胺,更优选链烷醇胺的碱,或其水溶液。
[0058]
在本发明的方法的一个优选实施方案中,步骤(d)中的乙氧基化在两个子步骤中进行,即(i)用每n-h官能最多1摩尔eo转化,随后(ii)在碱性催化下用更多环氧乙烷转化。
[0059]
本发明的进一步方面还有通过上述(和下面更详细描述的)本发明的方法可获得的如上文定义的两性改性的低聚丙烯亚胺乙氧基化物(a)。
[0060]
上文提到的本发明的方法的步骤(a)至(c)可如下进行。
[0061]
通过路线a:
[0062]
如先前在cn107311891中描述,可在反应容器中在5℃至80℃的温度下将1当量丙烯腈逐滴添加到任选溶解在溶剂中的过量的1,3-丙二胺、双-(3,3
’‑
氨基丙基)胺或双-(3,3
’‑
氨基丙基)-1,3-丙二胺或其混合物(最多100当量)中。在完全添加后,该反应可在所示温度下搅拌直至起始材料完全耗尽,然后冷却至室温。在任选纯化(但优选不纯化)后,粗制混合物可如先前在dd238043和/或jp08333308和/或wo2018046393中所述在压力反应器中在存在或不存在溶剂的情况下在升高的氢气和任选氨压力下发生由[cu]、[co]、[ni]、[pd]、[pt]或[ru]催化剂催化的氢化。在氢化过程中,温度可在70℃至200℃之间,优选在70℃至150℃之间,并且氢气压力在1至250巴之间,优选在50至250巴之间。可以例如通过过滤除去催化剂,并可在减压下除去挥发物。获得的所需低聚氨基化合物的混合物随后可在下一步骤中通过减压(《1巴)蒸馏分离,以得到纯化目标化合物双-(3,3
’‑
氨基丙基)胺、双-(3,3
’‑
氨基丙基)-1,3-丙二胺或三-(3,3’,3
”‑
氨基丙基)-1,3-丙二胺。通过路线b:
[0063]
如先前在cn102941160和/或wo9214709中描述,可在反应容器中在5℃至80℃的温度下将丙烯腈(最多2.5当量)逐滴添加到任选溶解在溶剂中的1当量的氨、1,3-丙二胺、双-(3,3
’‑
氨基丙基)胺或双-(3,3
’‑
氨基丙基)-1,3-丙二胺或其混合物中。在完全加成反应后,该反应可在所示温度下搅拌直至起始材料完全耗尽,然后冷却至室温。在任选纯化(但优选不纯化)后,粗制混合物可如先前在dd238043和/或jp08333308和/或wo 2018046393中所述在压力反应器中在存在或不存在溶剂的情况下在升高的氢气和任选氨压力下发生由[cu]、[co]、[ni]、[pd]、[pt]或[ru]催化剂催化的氢化。在氢化过程中,温度可在70℃至200℃之间,优选在70℃至150℃之间,并且氢气压力在1至250巴之间,优选在50至250巴之间。可以例如通过过滤除去催化剂,并可在减压下除去挥发物。获得的所需低聚氨基化合物的混合物随后可在下一步骤中通过减压(《1巴)蒸馏分离,以得到纯化目标化合物双-(3,3
’‑
氨基丙基)胺、双-(3,3
’‑
氨基丙基)-1,3-丙二胺或三-(3,3’,3
”‑
氨基丙基)-1,3-丙二胺。
[0064]
根据路线(a)或(b)的粗制混合物含有主要(》50摩尔%)线性低聚胺,优选多于70摩尔%线性低聚胺,更优选多于80摩尔%线性低聚胺。
[0065]
在本发明的一个优选实施方案中,根据路线(a)或(b)的粗制混合物通过蒸馏纯化,以分别除去来自单体、其它低聚物或支化结构和支化异构体的任何杂质,以获得纯度为至少80重量%,更优选至少90重量%,再更优选》95重量%的线性目标化合物双-(3,3
’‑
氨基丙基)胺、双-(3,3
’‑
氨基丙基)-1,3-丙二胺或三-(3,3’,3
”‑
氨基丙基)-1,3-丙二胺或其混合物。
[0066]
在本发明的一个实施方案中,纯化的双-(3,3
’‑
氨基丙基)-1,3-丙二胺或含有其的混合物可以10:1摩尔%或更高的比率含有异构化合物(根据式(ii)的在烷氧基化/改性
后的结构)。
[0067][0068]
其中
[0069]
r1相同或不同并选自c
1-c
4-烷基、h和自由电子对,其中所有r1的至少50%,优选至少80%,更优选至少90%是c
1-c
4-烷基,
[0070]
eo是指-ch
2-ch
2-o,
[0071]
y相同或不同并选自so
3-和h,其中所有y的至少30%,优选至少50%是so
3-,
[0072]
x相同或不同并选自5至50。
[0073]
上述制造方法的步骤(d)可如下进行。
[0074]
本发明的方法步骤(d)的特征在于仅使用环氧乙烷作为烷氧基化剂。其它环氧烷,如环氧丙烷对本发明的方法没有用并且不会得到本发明的产物及其在洗衣应用中的性质。
[0075]
在本发明的制造方法的步骤(d)的一个优选实施方案中,乙氧基化在两个步骤中进行。
[0076]
根据本发明的一个优选实施方案,在方法步骤(d)的第一步骤(i)中以低聚丙烯亚胺(ppi)的每nh基团0.2至1.0个环氧乙烷单元,优选低聚丙烯亚胺(ppi)的每nh基团0.5至0.99,更优选0.6至0.95个环氧乙烷单元,再更优选低聚丙烯亚胺(ppi)的每nh基团0.70至0.95个环氧乙烷单元的量加入环氧乙烷(欠羟乙基化(under-hydroxyethylation)。
[0077]
在本发明的方法的一个优选实施方案中,在步骤(i)中加入的每nh基团的环氧乙烷单元的最低量至少等同于稍后在步骤(ii)中加入的碱性催化剂c的量或更高,以防止在步骤(ii)的过程中通过催化剂c与环氧乙烷的直接反应形成聚乙二醇。
[0078]
优选地,在步骤(i)和(ii)中加入的环氧乙烷eo的量的总和在低聚丙烯亚胺(ppi)的每nh基团5至50个环氧乙烷单元,更优选低聚丙烯亚胺(ppi)的每nh基团10至40个环氧乙烷单元,再更优选低聚丙烯亚胺(ppi)的每nh基团15至30个环氧乙烷单元的范围内。
[0079]
优选地,方法步骤(d)的第一步骤(i)在不存在碱性催化剂的情况下进行。在方法步骤(d)的第一步骤(i)中可以加入水。
[0080]
在本发明的一个实施方案中,本发明的方法的步骤(d)的第二步骤(ii)在碱性催化剂存在下进行。合适的碱是例如lioh、naoh、koh、csoh及其混合物、钠或钾的醇盐,如甲醇钾(koch3)、叔丁醇钾、甲醇钠(naoch3)、正己醇钠和乙醇钠。催化剂的进一步实例是碱金属氢化物和碱土金属氢化物,如氢化钠和氢化钙,和碱金属碳酸盐,如碳酸钠和碳酸钾。优选的是碱金属氢氧化物,优选的是氢氧化钾和氢氧化钠,和碱金属醇盐,优选的是甲醇钾
(koch3)和甲醇钠(naoch3)。特别优选的是氢氧化钾和甲醇钾(koch3)。碱,例如koh的典型用量是相对于乙氧基化低聚丙烯亚胺(eppi)计0.02至10重量%,特别是0.05至1重量%。
[0081]
在本发明的方法的一个优选实施方案中,碱性催化剂c仅用于第二步骤(ii)并选自含碱土金属的碱性催化剂。
[0082]
本发明的方法中的碱性催化剂的一个尤其优选的实施方案是koh;koh可作为水溶液用于本发明的方法。
[0083]
在本发明的方法的一个实施方案中,碱性催化剂c以相对于乙氧基化低聚丙烯亚胺(eppi)计0.05至0.3重量%,优选0.15至0.25重量%的量添加。
[0084]
在一个实施方案中,在本发明的方法的步骤(d)的第一步骤(i)的过程中温度在90℃至160℃,优选100℃至150℃,更优选110℃至140℃的范围内。
[0085]
在一个实施方案中,在本发明的方法的步骤(d)的第二步骤(ii)的过程中温度在100℃至180℃,优选120℃至160℃,更优选120℃至145℃的范围内。
[0086]
在烷氧基化步骤的过程中比上文规定的温度高的温度也是可能的,但不优选,因为它们通常导致(更多)有色产物。
[0087]
在本发明的一个实施方案中,在本发明的方法的步骤(d)的第一步骤(i)的过程中的反应可在最多15巴,优选最多10巴,例如1至6巴的总压力下进行。用于进行该反应的优选容器是高压釜和管式反应器。
[0088]
在本发明的一个实施方案中,在本发明的方法的步骤(d)的第二步骤(ii)的过程中的反应可在最多15巴,优选最多10巴,例如2至10巴的总压力下进行。用于进行该反应的优选容器是高压釜和管式反应器。在本发明的方法的步骤(d)的第二步骤(ii)的过程中的反应可在不同中间体(即烷氧基化程度)处中断并可在追加或不追加催化剂的情况下继续。
[0089]
在本发明的方法的步骤(d)的步骤(ii)后获得的产物,即乙氧基化低聚丙烯亚胺(eppi)可用漂白剂处理。漂白剂优选选自硼酸盐、次氯酸盐、硼氢化物和过氧化氢。
[0090]
上文提到的本发明的方法的步骤(e):用硫酸二-c
1-c
4-烷基酯(至少部分)季铵化和转硫酸化。
[0091]
季铵化和转硫酸化步骤(e)是基于eppi的主链中的氨基的季铵化和聚环氧乙烷侧链的末端羟基部分的硫酸化的组合的方法。更一般而言,季铵化和转硫酸化步骤(e)包含形成硫酸化物类的子步骤(e1)(=季铵化)和提供一个或多个羟基部分的可控硫酸化的子步骤(e2)(=转硫酸化)。
[0092]
该方法的第一所需子步骤(e1)在碱性或接近ph中性条件下进行。该方法的第二子步骤(e2)在酸性条件下进行。
[0093]
下面详细描述该方法:
[0094]
子步骤(e1)(季铵化):在本发明的一个优选实施方案中,使0.5至1.0当量的硫酸化剂,优选0.8至0.99当量,最优选0.9至0.99当量与乙氧基化低聚丙烯亚胺的一个叔氨基反应,以形成低聚胺主链中的季铵离子和等量的硫酸化物类。如果需要,该方法可在溶剂存在下进行,优选可使用非反应性溶剂,如甲苯、甘醇二甲醚或二甘醇二甲醚。根据本发明的优选硫酸化剂是硫酸二烷基酯,优选硫酸二-c
1-c
4-烷基酯,更优选硫酸二-c
1-c
2-烷基酯,最优选硫酸二甲酯。本发明的方法的子步骤(e1)在碱性或接近ph中性条件下在0℃至180℃,优选40℃至100℃,再更优选50℃至90℃的温度下进行。该反应在放热时可通过任何合
适的手段控制,例如通过反应容器的冷却或通过提供回流冷凝器。
[0095]
子步骤(e2)(转硫酸化):硫酸化羟基物类的形成是本发明的方法的第二所需步骤。要硫化的每个羟基部分需要1当量的硫酸化物类。本发明的方法中的硫酸化物类的数目等同于低聚胺主链中的季铵离子的数目。根据转硫酸化步骤过程中的转化程度,在子步骤(e2)后获得的产物是两性改性的聚合物,(i)如果硫酸化物类在转硫酸化步骤的过程中完全(100%)转化,其净电荷为零(=中性聚合物),即低聚胺主链中的季铵离子和硫酸化羟基的数目相等;或(ii)如果在转硫酸化步骤的过程中仅部分(《100%)转化,其净电荷为正(=弱阳离子聚合物),即低聚胺主链中的季铵离子数略高于硫酸化羟基。为了控制转硫酸化步骤过程中的转化程度,配制人员可除去作为副产物形成的醇,优选c
1-c
4-醇,最优选甲醇(取决于步骤(e1)中所用的硫酸二-c
1-c
4-烷基酯的类型)。实际上,除去的醇副产物的相对量可用作控制转硫酸化步骤的转化程度的工具。可使用对配制人员方便的任何方法,例如蒸馏、吸收到分子筛中、结晶或沉淀,优选蒸馏。在许多情况下,优选已经在反应过程中除去副产物醇,优选通过蒸馏除去。
[0096]
根据本发明在转硫酸化步骤过程中的转化程度为至少50%,优选至少80%。因此,式(i)中的r1=c
1-c
4-烷基与y=so
3-的优选比率为平均1.0:1.0至1.0:0.8。
[0097]
子步骤(e2)后的最终产物作为内部两性离子获得,在转硫酸化步骤的过程中硫酸化物类不完全转化(《100%)的情况下具有可能额外的阳离子电荷。季铵化氮原子的抗衡离子是so
3-离子以致形成内部两性离子,和在转硫酸化步骤的过程中不完全转化的情况下可能额外的烷基硫酸根离子(c1至c4单烷基硫酸根),优选甲基硫酸根离子。
[0098]
子步骤(e2)必须在酸性条件下进行。合适的酸尤其是硫酸、盐酸、甲磺酸或路易斯酸(例如三氟化硼)。在根据本发明的方法中,优选使用硫酸。酸可以以足以形成所需产物的任何量添加,无论如何,本发明的方法在小于大约6,优选小于大约4,更优选小于大约3的ph下,最优选在大约2的ph下进行。实际上,相对于乙氧基化低聚丙烯亚胺(eppi)计大约0.01至1摩尔比的酸含量是优选的。催化剂可通过对配制人员方便的任何方式引入,无论如何,应该使用良好的混合。或者,可通过添加过量硫酸化剂并使这种过量试剂与有限的质子源,尤其是水反应而原位生成酸。本发明的方法的子步骤(e2)在0℃至200℃,优选40℃至150℃,再更优选70℃至120℃的温度下进行。该反应在放热时可通过任何合适的手段控制,例如通过反应容器的冷却或通过提供回流冷凝器。
[0099]
在本发明的优选实施方案中的硫酸的使用可能导致除羟基通过转硫酸化过程(即借助所用的硫酸二-c
1-c
4-烷基酯)转化成硫酸基团外,还在较小程度上作为副反应发生乙氧基化低聚丙烯亚胺的羟基的额外硫酸化。
[0100]
子步骤(e2)后的最终产物可进一步纯化以除去挥发性副产物和/或酸性催化剂,优选硫酸,或可作为混合物分离。可以例如通过蒸馏或在真空下汽提除去挥发性副产物,例如1,4-二氧杂环己烷。如果没有从子步骤(e2)后的最终产物中除去酸性催化剂,该混合物可以原样分离或可将酸性催化剂中和。在本发明的一个优选实施方案中,没有除去酸性催化剂,而是中和。任何合适的碱可用于中和酸性催化剂,尤其是氢氧化铵、氢氧化锂、氢氧化钠、氢氧化钾、氢氧化镁、氢氧化钙、氢氧化钡或胺。优选使用氢氧化锂、氢氧化钠、氢氧化钾或胺,再更优选氢氧化钠、链烷醇胺或其水溶液。
[0101]
在本发明的一个实施方案中,链烷醇胺用于中和酸性催化剂。在所用的酸是硫酸
的情况下,形成链烷醇胺的硫酸盐,并且如果使用碱的水溶液,最终在另外包含链烷醇胺的硫酸盐和水的混合物中获得本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)。
[0102]
子步骤(e2)后的最终产物,如果适用,在除去或中和酸性催化剂后,可以任何比率与水混合。优选地,子步骤(e2)后的最终产物与1至80重量%水,更优选1至60重量%水,再更优选5至50重量%水,最优选10至40重量%水混合,以降低粘度和改进操作。
[0103]
任选的后处理步骤还可包括调节最终产物的ph,尤其是如果该产物作为水溶液获得。任何合适的碱或酸可用于调节ph。优选地,使用氢氧化钠、氢氧化钾或胺作为碱;使用硫酸、盐酸或甲磺酸作为酸。在本发明的一个实施方案中,链烷醇胺用于调节ph。本发明的聚合物的水溶液的中性至微碱性ph是优选的以保护硫酸基团以免水解和裂解成游离羟基。因此,优选将最终产物在水中的ph调节到ph 6至14,更优选ph 6至11,再更优选ph 7至10。
[0104]
此外,可添加抗微生物剂以改善最终产物的水溶液的保存。
[0105]
下面是根据本发明的方法的非限制性实例:
[0106]
可将获自上述本发明的方法的步骤(d)的烷氧基化产物加热到升高的温度,通常在50℃至90℃之间,并在保护气氛(例如氮气气氛)下填充到反应器中。可将相对于乙氧基化低聚丙烯亚胺(eppi)摩尔过量的硫酸二-c
1-c
4-烷基酯,例如硫酸二甲酯(dms)优选在1小时至5小时的时间跨度内计量到反应器中以使反应器中的温度不超过90℃。然后该反应可在不超过90℃的温度下搅拌一段时间以完成反应,例如在1小时至5小时之间。
[0107]
然后可加入相对于乙氧基化低聚丙烯亚胺(eppi)计小于1当量的硫酸,并可将温度设定为70℃至120℃之间的值。可将反应器设定在真空下(例如5至30毫巴)1至6小时,任选在惰性气体汽提下。在反应完成后,可加入氢氧化钠或三乙醇胺(优选在水溶液中)和软化水,并可将ph调节到ph 7至10。然后可从反应器中除去液体产物。
[0108]
转硫酸化步骤(本发明的方法的步骤(e)的第二部分)也可类似于wo200424858a1(实施例4)中描述的方法进行。
[0109]
根据本发明的最终所得聚合物具有1000至20000g/mol,优选1500至15000g/mol,再更优选2000至10000g/mol的重均分子量(通过gpc测定,参见实验部分)。在本发明的一个优选实施方案中,两性改性的低聚丙烯亚胺乙氧基化物(a)具有2500至8000g/mol的重均分子量。
[0110]
包含本发明的化合物的洗衣制剂
[0111]
本发明的另一个方面还是一种洗衣制剂,其包含如上所述的本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)和/或本发明的化合物混合物的至少一种。
[0112]
因此,本发明的一个方面还是如上所述的本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)和/或本发明的化合物混合物在洗衣应用中的用途。
[0113]
根据本发明的洗衣制剂可以是液体、凝胶或固体组合物,固体实施方案包含例如粉末和片剂。液体组合物可包装为单位剂量。
[0114]
在本发明的一个实施方案中,两性改性的低聚丙烯亚胺乙氧基化物(a)是另外包含至少一种阴离子表面活性剂(b)的洗衣制剂(在本说明书中也称为洗衣护理组合物)的一种组分。
[0115]
合适的阴离子表面活性剂(b)的实例是c
8-c
12-烷基硫酸酯的碱金属和铵盐、c
12-c
18-脂肪醇醚硫酸酯的碱金属和铵盐、c
12-c
18-脂肪醇聚醚硫酸酯的碱金属和铵盐、乙氧基
化c
4-c
12-烷基酚(乙氧基化:3至50摩尔环氧乙烷/mol)的硫酸半酯的碱金属和铵盐、c
12-c
18-烷基磺酸的碱金属和铵盐、c
12-c
18
磺基脂肪酸烷基酯,例如c
12-c
18
磺基脂肪酸甲基酯的碱金属和铵盐,c
10-c
18-烷基芳基磺酸,优选n-c
10-c
18-烷基苯磺酸的碱金属和铵盐,c
10-c
18
烷基烷氧基羧酸酯的碱金属和铵盐,以及皂,例如c
8-c
24-羧酸的碱金属和铵盐。优选的是上述化合物的碱金属盐,特别优选钠盐。
[0116]
在本发明的一个实施方案中,阴离子表面活性剂(b)选自n-c
10-c
18-烷基苯磺酸和脂肪醇聚醚硫酸酯,其在本发明中特别是乙氧基化c
12-c
18-链烷醇(乙氧基化:1至50摩尔环氧乙烷/mol)的硫酸半酯,优选n-c
12-c
18-链烷醇的硫酸半酯。
[0117]
根据本发明的组合物可包含至少一种增洁剂(c)。在本发明中,没有区分增洁剂和在别处称为“助增洁剂(co-builder)”的组分。增洁剂(c)的实例是络合剂(下文也称为络合剂(c))、离子交换化合物和沉淀剂(c)。增洁剂选自柠檬酸盐、磷酸盐、硅酸盐、碳酸盐、膦酸盐、氨基羧酸盐和多羧酸盐。
[0118]
在本发明中,术语柠檬酸盐包括柠檬酸的单碱金属盐和二碱金属盐,特别是单钠盐和优选三钠盐,柠檬酸的铵盐或取代铵盐以及柠檬酸。柠檬酸盐可作为无水化合物或作为水合物,例如作为二水合柠檬酸钠使用。柠檬酸盐的量参考无水柠檬酸三钠计算。
[0119]
术语磷酸盐包括偏磷酸钠、正磷酸钠、磷酸氢钠、焦磷酸钠和多磷酸盐,如三聚磷酸钠。但是,优选地,根据本发明的组合物不含磷酸盐和多磷酸盐,其中包括磷酸氢盐,例如不含磷酸三钠、三聚磷酸五钠和偏磷酸六钠(“无磷酸盐”)。关于磷酸盐和多磷酸盐,“不含”在本发明中应理解为是指磷酸盐和多磷酸盐的含量总计为各自组合物的10重量ppm至0.2重量%,通过重量法测定。
[0120]
术语碳酸盐包括碱金属碳酸盐和碱金属碳酸氢盐,优选的是钠盐。特别优选的是na2co3。
[0121]
膦酸盐的实例是羟基烷烃膦酸盐和氨基烷烃膦酸盐。在羟基烷烃膦酸盐中,1-羟基乙烷-1,1-二膦酸盐(hedp)作为增洁剂特别重要。其优选作为钠盐使用,二钠盐是中性的,四钠盐是碱性的(ph 9)。合适的氨基烷烃膦酸盐优选是乙二胺四亚甲基膦酸盐(edtmp)、二亚乙基三胺五亚甲基膦酸盐(dtpmp)以及它们的更高级同系物。它们优选以中性反应钠盐的形式使用,例如作为edtmp的六钠盐或作为dtpmp的七钠和八钠盐。
[0122]
氨基羧酸盐和多羧酸盐的实例是次氮基三乙酸盐、乙二胺四乙酸盐、二亚乙基三胺五乙酸盐、三亚乙基四胺六乙酸盐、丙二胺四乙酸、乙醇-二甘氨酸、甲基甘氨酸二乙酸盐和谷氨酰胺二乙酸盐。术语氨基羧酸盐和多羧酸盐也包括它们各自的未取代或取代的铵盐和碱金属盐,如钠盐,特别是各自的完全中和化合物。
[0123]
硅酸盐在本发明中特别包括二硅酸钠和偏硅酸钠、硅铝酸盐,例如沸石和页硅酸盐,特别是式α-na2si2o5、β-na2si2o5和δ-na2si2o5的那些。
[0124]
根据本发明的组合物可含有选自上文未提到的材料的一种或多种增洁剂。增洁剂的实例是α-羟基丙酸和氧化淀粉。
[0125]
在本发明的一个实施方案中,增洁剂(c)选自多羧酸盐。术语“多羧酸盐”包括非聚合的多羧酸盐,如琥珀酸、c
2-c
16-烷基二琥珀酸盐、c
2-c
16-烯基二琥珀酸盐、乙二胺n,n
’‑
二琥珀酸、酒石酸二乙酸盐、碱金属丙二酸盐、酒石酸单乙酸盐、丙三羧酸、丁四羧酸和环戊烷四羧酸。
[0126]
低聚或聚合的多羧酸盐是例如聚天冬氨酸或特别是(甲基)丙烯酸均聚物或(甲基)丙烯酸共聚物的碱金属盐。
[0127]
合适的共聚单体是单烯属不饱和二羧酸,如马来酸、富马酸、马来酸酐、衣康酸和柠康酸。合适的聚合物特别是聚丙烯酸,其优选具有在2000至40 000g/mol,优选2000至10 000g/mol,特别是3000至8000g/mol的范围内的平均分子量mw。另外合适的共聚的聚羧酸盐特别是丙烯酸与甲基丙烯酸的共聚物和丙烯酸或甲基丙烯酸与马来酸和/或富马酸的共聚物。
[0128]
也可能使用选自单烯属不饱和c
3-c
10-单羧酸或c
4-c
10-二羧酸或其酐,如马来酸、马来酸酐、丙烯酸、甲基丙烯酸、富马酸、衣康酸和柠康酸的至少一种单体与至少一种如下所列的亲水或疏水改性共聚单体的共聚物。
[0129]
合适的疏水共聚单体是例如异丁烯、二异丁烯、丁烯、戊烯、己烯和苯乙烯、具有10个或更多个碳原子的烯烃或其混合物,例如1-癸烯、1-十二烯、1-十四烯、1-十六烯、1-十八烯、1-二十烯、1-二十二烯、1-二十四烯和1-二十六烯、c
22-α-烯烃、c
20-c
24-α-烯烃的混合物和具有每分子平均12至100个碳原子的聚异丁烯。
[0130]
合适的亲水共聚单体是具有磺酸酯或膦酸酯基团的单体,以及具有羟基官能或环氧烷基团的非离子单体。例如,可以提到:烯丙醇、异戊二烯醇、甲氧基聚乙二醇(甲基)丙烯酸酯、甲氧基聚丙二醇(甲基)丙烯酸酯、甲氧基聚丁二醇(甲基)丙烯酸酯、甲氧基聚(环氧丙烷-co-环氧乙烷)(甲基)丙烯酸酯、乙氧基聚乙二醇(甲基)丙烯酸酯、乙氧基聚丙二醇(甲基)丙烯酸酯、乙氧基聚丁二醇(甲基)丙烯酸酯和乙氧基聚(环氧丙烷-co-环氧乙烷)(甲基)丙烯酸酯。聚烷撑二醇在此可包含每分子3至50,特别是5至40,尤其是10至30个环氧烷单元。
[0131]
在此特别优选的含磺酸基团的单体是1-丙烯酰氨基-1-丙磺酸、2-丙烯酰氨基-2-丙磺酸、2-丙烯酰氨基-2-甲基丙磺酸、2-甲基丙烯酰氨基-2-甲基丙磺酸、3-甲基丙烯酰氨基-2-羟基丙磺酸、烯丙基磺酸、甲基烯丙基磺酸、烯丙氧基苯磺酸、甲基烯丙氧基苯磺酸、2-羟基-3-(2-丙烯氧基)丙磺酸、2-甲基-2-丙烯-1-磺酸、苯乙烯磺酸、乙烯基磺酸、丙烯酸3-磺丙酯、甲基丙烯酸2-磺乙酯、甲基丙烯酸3-磺丙酯、磺甲基丙烯酰胺、磺甲基甲基丙烯酰胺,和所述酸的盐,如其钠、钾或铵盐。
[0132]
特别优选的含膦酸酯基团的单体是乙烯基膦酸及其盐。
[0133]
此外,也可使用两性聚合物作为增洁剂。
[0134]
根据本发明的组合物可包含例如总共0.1至70重量%,优选10至50重量%,优选最多20重量%的增洁剂(c),尤其是在固体制剂的情况下。根据本发明的液体制剂优选包含0.1至8重量%的增洁剂(c)。
[0135]
根据本发明的制剂可包含一种或多种碱载体。如果需要碱性ph,碱载体确保例如至少9的ph。合适的是例如上文提到的碱金属碳酸盐、碱金属碳酸氢盐和碱金属偏硅酸盐,以及另外,碱金属氢氧化物。优选的碱金属在每种情况下是钾,特别优选的是钠。
[0136]
在本发明的一个实施方案中,根据本发明的洗衣制剂另外包含至少一种酶(d)。
[0137]
可用的酶是例如一种或多种脂肪酶、水解酶、淀粉酶、蛋白酶、纤维素酶、半纤维素酶、磷脂酶、酯酶、果胶酶、乳糖酶和过氧化物酶,和至少两种上述类型的组合。
[0138]
酶(d)可以足以提供有效清洁量的含量掺入。在根据本发明的洗涤剂组合物中,优
选的量在0.001重量%至5重量%活性酶的范围内。酶稳定剂体系,例如钙离子、硼酸、有机硼酸、丙二醇、短链羧酸和短链羧酸也可与酶一起使用。在本发明中,短链羧酸选自每分子具有1至3个碳原子的单羧酸和每分子具有2至6个碳原子的二羧酸。优选实例是甲酸、乙酸、丙酸、草酸、琥珀酸、hooc(ch2)3cooh、己二酸和上述至少两种的混合物,以及各自的钠盐和钾盐。
[0139]
根据本发明的组合物可包含一种或多种漂白剂(e)。
[0140]
优选的漂白剂(e)选自过硼酸钠——无水或例如作为一水合物或作为四水合物或所谓的二水合物,过碳酸钠——无水或例如作为一水合物,和过硫酸钠,其中术语“过硫酸盐”在每种情况下包括过酸h2so5的盐以及过氧二硫酸盐。
[0141]
在这方面,碱金属盐在每种情况下也可以是碱金属碳酸氢盐、碱金属过硼酸氢盐和碱金属过硫酸氢盐。但是,二碱金属盐在每种情况下是优选的。
[0142]
根据本发明的制剂可包含一种或多种漂白催化剂。漂白催化剂可选自oxaziridinium基漂白催化剂、增强漂白的过渡金属盐或过渡金属络合物,例如锰-、铁-、钴-、钌-或钼-salen络合物或羰基络合物。锰、铁、钴、钌、钼、钛、钒和铜与含氮的三脚配体的络合物以及钴-、铁-、铜-和钌-胺络合物也可用作漂白催化剂。
[0143]
根据本发明的制剂可包含一种或多种漂白活化剂,例如四乙酰乙二胺、四乙酰亚甲基二胺、四乙酰甘脲、四乙酰己二胺、酰化酚磺酸酯,例如正壬酰-或异壬酰氧基苯磺酸酯、n-甲基吗啉鎓-乙腈盐(“mma盐”)、三甲基铵乙腈盐,n-酰基酰亚胺,例如n-壬酰基琥珀酰亚胺、1,5-二乙酰基-2,2-二氧代六氢-1,3,5-三嗪(“dadht”)或腈季铵化合物(三甲基铵乙腈盐)。
[0144]
根据本发明的制剂可包含一种或多种缓蚀剂(f)。在本情况下,这被理解为包括抑制金属腐蚀的那些化合物。合适的缓蚀剂的实例是三唑,特别是苯并三唑、双苯并三唑、氨基三唑、烷基氨基三唑,以及酚衍生物,例如氢醌、邻苯二酚、羟基氢醌、没食子酸、间苯三酚或连苯三酚。
[0145]
在本发明的一个实施方案中,根据本发明的制剂包含总共0.1至1.5重量%的缓蚀剂。
[0146]
根据本发明的制剂还可包含附加清洁聚合物(g)或去污聚合物(h)。附加清洁聚合物(g)可包括但不限于,多官能聚乙烯亚胺(例如basf’shp20)和/或多官能二胺(例如basf’shp96)。
[0147]
包含本发明的两性改性的低聚丙烯亚胺乙氧基化物(a)的洗衣制剂还可包含至少一种抗微生物剂(i)。
[0148]
抗微生物剂可选自2-苯氧基乙醇(cas-no.122-99-6,例如可获自basf的pe)或4,4
’‑
二氯-2-羟基二苯醚(cas:3380-30-1)及其组合。
[0149]
4,4
’‑
二氯-2-羟基二苯醚可作为溶液使用,例如30重量%的4,4
’‑
二氯-2-羟基二苯醚在1,2-丙二醇中的溶液,例如可获自basf的hp 100。
[0150]
本发明的洗衣制剂可包含上述名单中的至少一种抗微生物剂和/或其组合和/或与至少一种在此未列举的其它抗微生物剂的组合。
[0151]
抗微生物剂可以相对于聚合物的总重量计0.0001至10%的浓度添加到本发明的
洗衣制剂中。
[0152]
优选地,该制剂含有0.01%至5%,更优选0.1%至2%的浓度的2-苯氧基乙醇和/或0.001%至1%,更优选0.002%至0.6%的浓度的4,4
’‑
二氯-2-羟基二苯醚(在所有情况下相对于聚合物的总重量计)。
[0153]
根据本发明的制剂可包含选自非离子表面活性剂和两性表面活性剂的至少一种附加表面活性剂。
[0154]
非离子表面活性剂
[0155]
(附加)表面活性剂的实例特别是非离子表面活性剂。优选的非离子表面活性剂是烷氧基化醇和烷氧基化脂肪醇、环氧乙烷和环氧丙烷的二嵌段和多嵌段共聚物、和山梨糖醇酐与环氧乙烷或环氧丙烷的反应产物,还有烷基酚乙氧基化物、烷基糖苷、聚羟基脂肪酸酰胺(葡糖酰胺)和所谓的胺氧化物。
[0156]
烷氧基化醇和烷氧基化脂肪醇的优选实例是例如通式(iii)的化合物
[0157][0158]
其中变量如下定义:
[0159]
r1选自直链c
1-c
10-烷基,优选乙基,特别优选甲基,
[0160]
r2选自c
8-c
22-烷基,例如n-c8h
17
、n-c
10h21
、n-c
12h25
、n-c
14h29
、n-c
16h33
或n-c
18h37
,
[0161]
r3选自c
1-c
10-烷基、甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、仲戊基、新戊基、1,2-二甲基丙基、异戊基、正己基、异己基、仲己基、正庚基、正辛基、2-乙基己基、正壬基、正癸基或异癸基,m和n在0至300的范围内,其中n和m之和为至少1。优选地,m在1至100的范围内且n在0至30的范围内。
[0162]
在此,通式(iii)的化合物可以是嵌段共聚物或无规共聚物,优选的是嵌段共聚物。
[0163]
烷氧基化醇和烷氧基化脂肪醇的其它优选实例是例如通式(iv)的化合物
[0164][0165]
其中变量如下定义:
[0166]
r1相同或不同并选自直链c
1-c
4-烷基,优选在每种情况下相同并且是乙基,特别优选甲基,
[0167]
r4选自c
6-c
20-烷基,特别是n-c8h
17
、n-c
10h21
、n-c
12h25
、n-c
14h29
、n-c
16h33
、n-c
18h37
,
[0168]
a是在0至6,优选1至6的范围内的数,
[0169]
b是在0至20,优选4至20的范围内的数,
[0170]
d是在4至25的范围内的数。
[0171]
优选地,a和b的至少一个大于0。
[0172]
在此,通式(iv)的化合物可以是嵌段共聚物或无规共聚物,优选的是嵌段共聚物。
[0173]
进一步合适的非离子表面活性剂选自由环氧乙烷和环氧丙烷组成的二嵌段和多嵌段共聚物。进一步合适的非离子表面活性剂选自乙氧基化或丙氧基化山梨糖醇酐酯。胺氧化物,如月桂基二甲基胺氧化物(“月桂胺氧化物”)或烷基酚乙氧基化物或烷基聚糖苷或聚羟基脂肪酸酰胺(葡糖酰胺)同样合适。合适的其它非离子表面活性剂的综述可见于ep-a 0 851 023和de-a 198 19 187。
[0174]
也可存在两种或更多种不同的非离子表面活性剂的混合物。
[0175]
两性表面活性剂的实例是c
12-c
18-烷基甜菜碱和磺基甜菜碱。
[0176]
根据本发明的制剂还可包含水和附加有机溶剂,例如乙醇或丙二醇。
[0177]
附加任选成分可以是,但不限于粘度改进剂、阳离子表面活性剂、泡沫促进或泡沫减少剂、香精、染料、荧光增白剂和染料转移抑制剂。
实施例
[0178]
在以下段落中,给出几个实验实施例以例示本发明的一些方面。本发明的实施例和对比例的合成:
[0179]
低聚胺的合成
[0180]
双-(3,3
’‑
氨基丙基)胺(二亚丙基三胺,dpta)的合成
[0181]
在反应容器中在60℃下将丙烯腈(7.8kg,0.15kmol,1.0当量)逐滴引入过量的1,3-二氨基丙烷(27.0kg,0.36kmol,2.5当量)中并保持在65℃下。在完全加成反应后,该反应在60℃下搅拌2小时,然后冷却至室温。粗制混合物接着通过gc色谱法分析并发现得到45%(gc面积-%)未反应起始材料、47%(gc面积-%)所需单氰乙基化化合物和7%(gc面积-%)二氰乙基化化合物(34.8kg)的分布。随后并且未经任何进一步纯化,上述粗制混合物在由[co]-催化剂催化的固定床压力反应器中在90℃和200巴氢气压力下与氨(28-45当量)一起发生氢化。粗制低聚胺混合物在减压(140至20毫巴)下和在升高的温度(120-220℃柱温)下进行分馏以得到作为无色液体的dpta(134℃;20毫巴;纯度》99%)。
[0182]
gc分析(30m rtx5 amin柱;注入温度在60℃,然后以10℃/min加热至280℃):r
t
=11.39min(dpta)和r
t
=17.25min(tpta)。
[0183]1h-nmr(500mhz,cdcl3):):
[0184]
13
c-nmr(125mhz,cdcl3):双-(3,3
’‑
氨基丙基)-1,3-丙二胺(三亚丙基四胺,tpta)的合成
[0185]
在反应容器中在13℃下将丙烯腈(795g,15.0mol,2.05当量)在4小时内逐滴引入1,3-二氨基丙烷(542g,7.3mol,1.0当量)中并保持低于15℃。在完全加成反应后,该反应在15℃下搅拌另外2小时,然后升温至室温。随后并且未经任何进一步纯化,上述粗制混合物在由雷尼镍催化剂(5重量%)催化的分批压力反应器中在100℃和200巴氢气压力下发生氢化并搅拌12小时。在完全反应后,通过用氮气吹扫反应容器而猝灭反应,通过过滤除去催化剂并在减压下除去挥发物。在减压(3毫巴)下和在升高的温度(170℃柱温)下蒸馏后获得所需目标化合物并得到作为无色液体的tpta(130℃;3毫巴;》99%纯度)。
[0186]
gc分析(30m rtx5 amin柱;注入温度在60℃,然后以10℃/min加热至280℃):r
t
=
17.25min(tpta)。
[0187]1h-nmr(500mhz,meod):nmr(500mhz,meod):
[0188]
13
c-nmr(125mhz,meod):nmr(125mhz,meod):
[0189]
三-(3,3’,3
”‑
氨基丙基)-1,3-丙二胺(四亚丙基五胺,tppa)的合成
[0190]
在反应容器中在50℃下将丙烯腈(339g,6.4mol,2.0当量)在3小时内逐滴引入三亚丙基四胺(tpta,598g,3.2mol,1.0当量)在thf(750ml)中的混合物中。在完全加成反应后,该反应在50℃下搅拌另外2小时,然后冷却至室温。随后并且未经任何进一步纯化,上述粗制混合物在由雷尼钴催化剂(5重量%)/thf催化的分批压力反应器中在120℃和100巴氢气压力下发生氢化并搅拌8小时。在完全反应后,通过用氮气吹扫反应容器而猝灭反应,通过过滤除去催化剂并在减压下除去溶剂。在减压(2毫巴)下和在升高的温度(270℃柱温)下蒸馏后与五亚丙基六胺(ppha)一起获得所需目标化合物并得到作为黄色油的tppa(147℃;2毫巴;93%纯度)。
[0191]
gc分析(30m rtx5 amin柱;注入温度在80℃,然后以15℃/min加热至280℃):r
t
=20.23min(tppa)。
[0192]
聚合物p1(本发明)
[0193]
将96.03克二亚丙基三胺(dpta,0.83摩尔,1当量)和10克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至100℃并将130克环氧乙烷(2.95摩尔,3.56当量)在7小时内计量到反应器中。此后,将反应混合物保持在100℃以进行后反应。在真空下除去挥发性化合物并从反应器中取出221.5克清澈的高粘性产物。
[0194]
将39.8克先前获得的产物填充到钢压力反应器中并加入2.4克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至120℃并在6小时内加入548克环氧乙烷(12.4摩尔,99.7当量)。在真空下除去挥发性化合物并获得589克棕色固体。
[0195]
将200克所得乙氧基化物(0.044摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将16.2克硫酸二甲酯(0.13摩尔,2.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.6克硫酸(0.036摩尔,0.9当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入5.4克氢氧化钠(50%水溶液)和40克软化水,并从反应器中取出橙色液体产物。
[0196]
聚合物p2(本发明)
[0197]
将297.9克三亚丙基四胺(tpta,1.58摩尔,1当量)和29.8克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至100℃并将335克环氧乙烷(7.61摩尔,4.81当量)在10小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出626.4克清澈的高粘性产物。
[0198]
将100克先前获得的产物填充到钢压力反应器中并加入5.5克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定2巴的氮气压力。将反应器加热至120℃并在16小时内加入1270克环氧乙烷(28.8摩尔,115.2当量)。在真空下除去挥发性化合物并获得1374.2克棕色固体。
[0199]
将705.1克所得乙氧基化物(0.13摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将62.1克硫酸二甲酯(0.49摩尔,3.8当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将8.0克硫酸(0.08摩尔,0.6当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入11.0克氢氧化钠(50%水溶液)和650克软化水,并从反应器中取出橙色液体产物。
[0200]
聚合物p3(本发明)
[0201]
将138.9克三亚丙基四胺(tpta,0.74摩尔,1当量)和13.9克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至100℃并将156克环氧乙烷(3.54摩尔,4.81当量)在10小时内计量到反应器中。此后,将反应混合物保持在100℃5小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出290克清澈的高粘性产物。
[0202]
将63克先前获得的产物填充到钢压力反应器中并加入3.0克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至120℃并在10小时内加入696克环氧乙烷(15.8摩尔,100.3当量)。在真空下除去挥发性化合物并获得754.8克棕色固体。
[0203]
将556克所得乙氧基化物(0.12摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将57.4克硫酸二甲酯(0.49摩尔,3.8当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将7.0克硫酸(0.07摩尔,0.6当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入10.0克氢氧化钠(50%水溶液)和500克软化水,并从反应器中取出橙色液体产物。
[0204]
聚合物p4(本发明)
[0205]
将173.8克三亚丙基四胺(tpta,0.92摩尔,1当量)和17.3克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至100℃并将195克环氧乙烷(4.43摩尔,4.81当量)在10小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出366.8克清澈的高粘性产物。
[0206]
将60克先前获得的产物填充到钢压力反应器中并加入4.9克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至120℃并在15小时内加入1159克环氧乙烷(26.2摩尔,174.6当量)。在真空下除去挥发性化合物并获得1233克棕色固体。
[0207]
将488.1克所得乙氧基化物(0.06摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将29.3克硫酸二甲酯(0.23摩尔,3.87当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小
时。将6.7克硫酸(0.07摩尔,0.6当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入8.5克氢氧化钠(50%水溶液)和488.1克软化水,并从反应器中取出橙色液体产物。
[0208]
聚合物p5(本发明)
[0209]
将83.3克三亚丙基四胺(tpta,0.44摩尔,1当量)和8.3克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至120℃并将93.5克环氧乙烷(2.12摩尔,4.83当量)以内部压力不超过5.5巴的方式计量到反应器中。此后,将反应混合物保持在120℃6小时以进行后反应。加入9.1克氢氧化钾(50%水溶液)并在减压下除去水。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至140℃并以内部压力不超过5.5巴的方式加入844克环氧乙烷(19.2摩尔,43.6当量)。使该混合物后反应6小时。在真空下除去挥发性化合物并获得952.2克棕色粘性液体。
[0210]
将494克先前获得的烷氧基化物装载到钢压力反应器中,用氮气惰化,加热至140℃。设定2.5巴的氮气预压力并将667.4克环氧乙烷(15.15摩尔,34.4当量)以内部压力保持低于5.5巴的方式添加到反应器中。使该混合物后反应6小时。在真空中除去挥发性化合物并作为产物获得1060.8克棕色固体。
[0211]
将326.3克所得乙氧基化物(0.06摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将29.9克硫酸二甲酯(0.24摩尔,3.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将4.0克硫酸(0.04摩尔,0.68当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入9.14克三乙醇胺和143.1克软化水,并从反应器中取出橙色液体产物。
[0212]
聚合物p6(本发明)
[0213]
将62.9克四亚丙基五胺(tppa,0.26摩尔,1当量)和6.3克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定3.5巴的氮气压力。将反应器加热至100℃并将60克环氧乙烷(1.36摩尔,5.2当量)在7小时内计量到反应器中。此后,将反应混合物保持在100℃以进行后反应。在真空下除去挥发性化合物并加入6.2克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1.5巴的氮气压力。将反应器加热至120℃并在12小时内加入1435克环氧乙烷(32.575摩尔,125当量)。在真空下除去挥发性化合物并获得1589.2克棕色固体。
[0214]
将314.2克所得乙氧基化物(0.05摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将31.4克硫酸二甲酯(0.25摩尔,4.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.5克硫酸(0.036摩尔,0.7当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入5.0克氢氧化钠(50%水溶液)和300克软化水,并从反应器中取出橙色液体产物。
[0215]
对比例:
[0216]
聚合物cp1(对比)
[0217]
将297.9克三亚丙基四胺(tpta,1.58摩尔,1当量)和29.8克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至100℃并
将335克环氧乙烷(7.61摩尔,4.81当量)在10小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出626.4克清澈的高粘性产物。
[0218]
将100克先前获得的产物填充到钢压力反应器中并加入5.5克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定2巴的氮气压力。将反应器加热至120℃并在16小时内加入1270克环氧乙烷(28.8摩尔,115.2当量)。在真空下除去挥发性化合物并获得1374.2克棕色固体。
[0219]
聚合物cp2(对比)
[0220]
将99.1克三亚丙基四胺(tpta,0.53摩尔,1当量)和9.9克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1.0巴的氮气压力。将反应器加热至100℃并将112克环氧乙烷(2.54摩尔,4.83当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出210克清澈的高粘性产物。
[0221]
将39.2克先前获得的产物填充到钢压力反应器中并加入1.1克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至120℃并在10小时内加入498克环氧乙烷(11.3摩尔,115.2当量)。在真空下除去挥发性化合物并获得536克棕色固体。
[0222]
将115克所得乙氧基化物(0.02摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将10.3克硫酸二甲酯(0.08摩尔,3.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。加入氢氧化钠(50%水溶液)以将ph设定至8.2。作为浅棕色固体获得产物。
[0223]
聚合物cp3(对比)
[0224]
将500克聚丙烯亚胺和17克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至120℃并将348克环氧乙烷在6小时内计量到反应器中。此后,将反应混合物保持在120℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出825克黄色高粘性产物。
[0225]
将90克先前获得的产物填充到钢压力反应器中并加入3.5克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定2巴的氮气压力。将反应器加热至120℃并在16小时内加入783克环氧乙烷(17.8摩尔)。在真空下除去挥发性化合物并获得875克棕色固体。
[0226]
聚合物cp4(对比)
[0227]
将500克聚丙烯亚胺和17克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至120℃并将348克环氧乙烷在6小时内计量到反应器中。此后,将反应混合物保持在120℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出825克黄色高粘性产物。
[0228]
将90克先前获得的产物填充到钢压力反应器中并加入3.5克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定2巴的氮气压力。将反应器加热至120℃并在16小时内加入783克环氧乙烷(17.8摩尔)。在真空下除去挥发性化合物并获得875克棕色固体。
[0229]
将78.1克所得乙氧基化物加热至60℃并在氮气气氛下填充到玻璃反应器中。将6.6克硫酸二甲酯(0.05摩尔)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时并用5.4克氢氧化钠(50%水溶液)中和,获得82.2克棕色固体。
[0230]
将33.0克棕色固体加热至60℃并将1.2克硫酸添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入2.7克氢氧化钠(50%水溶液)。作为棕色固体获得产物。
[0231]
聚合物cp5(对比)
[0232]
将98.9克1,3-丙二胺(1,3-pda,1.33摩尔,1当量)和9.9克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1.0巴的氮气压力。将反应器加热至100℃并将189克环氧乙烷(4.29摩尔,3.23当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出210克清澈的高粘性产物。
[0233]
将50.05克先前获得的产物填充到钢压力反应器中并加入3.3克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至120℃并在10小时内加入788克环氧乙烷(17.9摩尔,76.9当量)。在真空下除去挥发性化合物并获得838.1克棕色固体。
[0234]
将200克所得乙氧基化物(0.06摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将13.8克硫酸二甲酯(0.11摩尔,1.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.6克硫酸(0.04摩尔,0.6当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入5.0克氢氧化钠(50%水溶液)和40克软化水,并从反应器中取出橙色液体产物。
[0235]
聚合物cp6(对比)
[0236]
将364克己二胺(hmda,3.13摩尔,1当量)和36.4克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.0巴的氮气压力。将反应器加热至100℃并将442克环氧乙烷(10.0摩尔,3.19当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出795.2克清澈的高粘性产物。
[0237]
将80克(0.43摩尔,1.0当量)先前获得的产物填充到钢压力反应器中并加入3.3克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至130℃并在15小时内加入1053克环氧乙烷(23.9摩尔,55.7当量)。在真空下除去挥发性化合物并获得1149.4克棕色固体。
[0238]
将364克所得乙氧基化物(0.1摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将24.8克硫酸二甲酯(0.20摩尔,1.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.4克硫酸(0.03摩尔,0.3当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入3.27克氢氧化钠(50%水溶液)和384克软化水,并从反应器中取出液体产物。
[0239]
聚合物cp7(对比)
[0240]
将97.9克乙二胺(eda,1.63摩尔,1当量)和9.7克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1.0巴的氮气压力。将反应器加热至100℃并将230克环氧乙烷(5.22摩尔,3.2当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出327克清澈的高粘性产物。
[0241]
将42.6克(0.21摩尔,1.0当量)先前获得的产物填充到钢压力反应器中并加入3.0克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至130℃并在15小时内加入717克环氧乙烷(16.3摩尔,77.5当量)。在真空下除去挥发性化合物并获得752.8克棕色固体。
[0242]
将200克所得乙氧基化物(0.06摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将13.9克硫酸二甲酯(0.11摩尔,1.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将4.2克硫酸(0.04摩尔,0.6当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入7.8克氢氧化钠(50%水溶液)和40克软化水,并从反应器中取出橙色液体产物。
[0243]
聚合物cp8(对比)
[0244]
将96.7克二亚乙基二胺(deta,0.94摩尔,1当量)和9.7克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1.0巴的氮气压力。将反应器加热至100℃并将136克环氧乙烷(3.08摩尔,3.3当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出231克清澈的高粘性产物。
[0245]
将45.9克(0.16摩尔,1.0当量)先前获得的产物填充到钢压力反应器中并加入2.9克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至130℃并在15小时内加入696克环氧乙烷(15.8摩尔,98.8当量)。在真空下除去挥发性化合物并获得732.7克棕色固体。
[0246]
将200克所得乙氧基化物(0.04摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将16.5克硫酸二甲酯(0.13摩尔,2.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.9克硫酸(0.04摩尔,0.8当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入6.8克氢氧化钠(50%水溶液)和40克软化水,并从反应器中取出橙色液体产物。
[0247]
聚合物cp9(对比)
[0248]
将233.6克三亚乙基四胺(teta,1.60摩尔,1当量)和23.3克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定1.0巴的氮气压力。将反应器加热至100℃并将338克环氧乙烷(7.67摩尔,4.8当量)在6小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出571克清澈的高粘性产物。
[0249]
将46.3克(0.13摩尔,1.0当量)先前获得的产物填充到钢压力反应器中并加入2.8
克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定1巴的氮气压力。将反应器加热至130℃并在15小时内加入658克环氧乙烷(14.9摩尔,114.9当量)。在真空下除去挥发性化合物并获得694克棕色固体。
[0250]
将200克所得乙氧基化物(0.04摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将17.4克硫酸二甲酯(0.14摩尔,3.75当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将3.0克硫酸(0.03摩尔,0.8当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入7.6克氢氧化钠(50%水溶液)和40克软化水,并从反应器中取出粘性液体产物。
[0251]
聚合物cp10(对比)
[0252]
pei600+20eo/nh,如wo9532272或us9738754中所述合成。
[0253]
聚合物cp11(对比)
[0254]
将400克三亚丙基四胺(tpta,2.12摩尔,1当量)和40克水装载到钢压力反应器中。该反应器用氮气吹扫以除去空气并设定2.5巴的氮气压力。将反应器加热至100℃并将450克环氧乙烷(10.22摩尔,4.8当量)在10小时内计量到反应器中。此后,将反应混合物保持在100℃6小时以进行后反应。在真空下除去挥发性化合物并从反应器中取出945克清澈的高粘性产物。
[0255]
将50.0克(0.13摩尔,1.0当量)先前获得的产物填充到钢压力反应器中并加入3.0克氢氧化钾(50%水溶液)。在减压下除去水。该反应器用氮气吹扫以除去空气并设定2巴的氮气压力。将反应器加热至130℃并在6小时内加入337克环氧乙烷(7.65摩尔,61.1当量)。使该混合物后反应6小时。此后,将87克环氧丙烷(1.50摩尔,12.0当量)在2小时内计量到反应器中。使混合物在130℃下后反应6小时。随后,将264克环氧乙烷(5.99摩尔,48.0当量)在130℃下计量到反应器中并使该混合物后反应6小时。在真空下除去挥发性化合物并获得755克黄色粘性液体。
[0256]
将451.6克所得乙氧基化物(0.08摩尔,1当量)加热至60℃并在氮气气氛下填充到玻璃反应器中。将39.1克硫酸二甲酯(0.31摩尔,3.9当量)以每分钟加入1毫升dms的方式计量到反应器中。在添加时,温度提高到70℃。在添加完成后,使混合物在70℃下后反应2小时。将5.80克硫酸(0.06摩尔,0.7当量)添加到反应器中,将温度提高到90℃并将反应器设定在真空(15毫巴)下3小时。在反应完成后,加入7.9克氢氧化钠(50%水溶液)和440克软化水,并从反应器中取出橙色液体产物。
[0257]
聚合物cp12(对比)
[0258]
pei2000+32.5eo/nh,如wo2020/030469(聚合物p.2)中所述合成:
[0259]
根据us 2010/0216949中描述的程序在2升高压釜中装载508.5克完全脱水且无co2的pei2000。然后,通过添加89克h2o,将pei2000制成85重量%水溶液。该容器用最多5巴的氮气压力吹扫3次,最后用2巴氮气垫使容器惰性化。将温度平衡在100℃,随后经6小时计量加入261克环氧乙烷并使其进一步反应1小时。产物pei2000+0.5eo/nh然后用氮气吹扫以汽提任何残留eo,从反应器中排空,并汽提出水和任何残留eo。将53.0克该材料装载到干净的空置2升高压釜中。然后计量加入4.8克50重量%koh水溶液并与pei2000+0.5eo/nh一起搅拌。随后,在120℃下在10毫巴下从混合物中汽提出水2小时。然后将温度提高到130℃,用
2巴氮气垫使容器惰性化,在大约3.5巴(初始压力)至大约8巴(在eo计量结束时的压力)的总压力下经12小时计量加入1150克环氧乙烷,并使其后反应至少6小时。样品然后用氮气吹扫以汽提任何残留eo,从反应器中排空,并在真空(20毫巴,90℃)中汽提出水和任何残留eo。获得1223克淡棕黄色固体。
[0260]
聚合物的表征
[0261]
通过凝胶渗透色谱法(gpc)测定分子量。所用条件是使用六氟异丙醇和0.05%三氟乙酸钾盐作为溶剂。将柱温箱温度设定为35℃,流量为1ml/min。注入50μl样品,并将样品浓度设定为1.5mg/ml。在聚合物溶解后使用millipore millflex fg(0.2μm)过滤器过滤样品以避免柱堵塞。使用下列柱:hfip保护柱(直径:8mm,长度5cm)、pl hfip凝胶柱(分离材料苯乙烯-二乙烯基苯,直径:7.5mm,长度:30cm)和pl hfipgel柱(分离材料苯乙烯-二乙烯基苯,直径:7.5mm,长度:30cm,排阻尺寸:100
–
100000g/mol)。gpc系统使用在800至2200000g/mol的分子量范围内的pmma标样校准。使用折射率(ri)检测器(dri agilent 1000)检测洗出液。
[0262]
本发明的聚合物和对比聚合物的分析数据概括在表1中。
[0263]
表1.本发明的两性改性的低聚丙烯亚胺乙氧基化物和对比聚合物的组成和物理化学表征
[0264][0265]
*malls检测器
[0266]
cp.1和cp.2:类似于本发明的聚合物(即基于低聚丙烯亚胺)但没有两性改性的聚合物。
[0267]
cp.3和cp.4:ep2961821中描述的聚合物。
[0268]
cp.6(和基于不同胺的类似聚合物cp.5和cp.7):描述在wo200424858中。
[0269]
cp.8和cp.9:类似于本发明的聚合物(即基于低聚丙烯亚胺)但基于低聚乙烯亚胺的聚合物。
[0270]
cp.10:pei乙氧基化物,如ep112593(应用)、wo9532272或us9738754(合成)中所述。
[0271]
cp.11:含有混合eo/po烷氧基化物链的聚合物,类似于ep3039109和ep3039057中描述的结构。
[0272]
cp.12:具有高分子量的pei乙氧基化物,如wo2020/030469(聚合物p.2)中所述。
[0273]
应用实验
[0274]
初次清洗性能
[0275]
为了测定初次去污力,通过使用反射计(datacolor sf600 plus)测定洗涤后的污渍与未沾污的白色织物之间的颜色差异(δe),测量对聚酯织物(warwick equest,consett,uk)上的圆形红色陶土和黄色陶土污渍的清洁性能。在1个实验中使用4个圆形红色陶土污渍和4个黄色陶土污渍(即2片聚酯试验织物,各含2个圆形红色陶土污渍和2个黄色陶土污渍),各实验重复3次,因此红色陶土和黄色陶土在每种试验条件下都获得总共12个洗过的污渍以计算平均δe值。通过使用这些δe值,计算所谓的“标准化清洁性能”(δδe)。“标准化清洁性能”(δδe)是包括相应的两性改性的低聚丙烯亚胺乙氧基化物或对比聚合物的衣物洗涤剂vs.不含任何两性改性的低聚丙烯亚胺乙氧基化物或对比聚合物的衣物洗涤剂的性能差异。
[0276]
表2显示衣物洗涤剂的组成。表3显示洗涤试验条件且表4概括所得标准化清洁性能。表4中所示的标准化清洁性能是对红色和黄色陶土的标准化清洁性能的总和。δδe值的总和越大,相应的两性改性的低聚丙烯亚胺乙氧基化物或对比聚合物对清洁性能的正面贡献越大。
[0277]
表2.液体衣物洗涤剂的组成
[0278][0279]
*)所有数据是重量%活性成分,与各自的产品形式无关。
[0280]
表3.用于评估初次去污力的洗涤条件
[0281][0282]
*)在洗涤实验后,在用反射计测量之前,试验织物用12
°
fh水冲洗(2次),然后在环境室温下干燥整夜。
[0283]
表4.来自洗涤试验的结果
[0284][0285]
*)所有数据是重量%活性成分,与各自的产品形式无关。
[0286]
**)δδe总和的所用方法的95%置信区间是+/-1.5。
[0287]
注:对比聚合物cp.10和cp.12(都基于pei核)表现出明显比本发明的聚合物差的性能,即使在三倍高的浓度水平下也是如此。在0.83重量%的较低浓度水平下,对cp.12完全无法检测到显著清洁性能。
[0288]
来自洗涤试验的结果清楚证明与现有技术中描述的对比聚合物相比本发明的聚
合物的优越性能。除cp.2和cp.11外的所有对比聚合物表现出明显比本发明的聚合物差的性能。如果考虑到所用方法的95%置信区间,对比聚合物cp.2表现出与本发明的聚合物相比定向较差的性能。
[0289]
粘度
[0290]
为了测定聚合物对液体洗衣制剂的粘度的影响,在每种情况下将0.5重量%或1.45重量%的两性改性的低聚丙烯亚胺乙氧基化物或对比聚合物分别配制到含有0.85重量%的固定含量的hase增稠聚合物的液体洗涤剂中(制剂f.1)。用50重量%naoh水溶液将ph调节至ph 7.5。将制剂用磁搅拌器搅拌2小时,随后在没有机械搅拌的情况下储存另外24小时。然后使用旋转流变仪rheolab qc(anton paar,ostfildern,germany)用心轴cc27在室温(23℃)下测量样品的粘度。在0至1200 1/s剪切速率下进行测量。
[0291]
表5显示最终制剂的组成,表6概括在20 1/s剪切速率下获得的粘度。
[0292]
表5.液体洗衣制剂的组成
[0293][0294]
*)所有数据是重量%活性成分,与各自的产品形式无关。
[0295]
表6.液体洗衣制剂的粘度
[0296][0297]
*)所有数据是重量%活性成分,与各自的产品形式无关。
[0298]
**)所用方法的线性标准偏差为+/-10mpa*s,由3种相同制剂的测量得出。
[0299]
注:类似于洗涤试验(见表4),使用与所有其它聚合物(本发明的聚合物和对比聚合物)相比3倍高的浓度水平的pei乙氧基化物cp.10和cp.12。
[0300]
来自粘度测量的结果清楚证实了本发明的聚合物的优越性:与所研究的对比聚合物相比(对比聚合物cp.12除外,但是,由于其清洁性能极差(见表4),这种聚合物不符合本发明的聚合物的目标标准),尤其是与对比聚合物cp.2和cp.11(它们已表现出相当的的清洁性能(cp.11)或仅稍差的清洁性能(cp.2)(见表4))相比,本发明的聚合物使得液体衣物洗涤剂的粘度明显更高。
[0301]
来自表4(清洁性能)和表6(液体衣物洗涤剂的粘度)的结果的组合清楚证明只有本发明的聚合物带来优异清洁性能(尤其是在低浓度下,优选在液体衣物洗涤剂中《1重量%)和洗涤剂制剂的高粘度的所需目标状况。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1