用于癌症免疫疗法的综合生物标志物的制作方法
用于癌症免疫疗法的综合生物标志物
相关申请的交叉引用
1.本技术要求2020年4月29日提交的美国临时专利申请第63/017,542号和2020年6月18日提交的美国临时专利申请第63/040,943号的优先权。通过引用将每个申请的全部内容并入本文用于所有目的。领域
2.本公开大体上涉及基于来自生物样品的基因组和转录物组度量来确定综合生物标志物的系统和方法。更具体地,但不是以限制的方式,本公开涉及基于基因组和转录物组度量来确定综合生物标志物得分,所述综合生物标志物得分鉴定受试者对特定类型的免疫疗法治疗的的响应性的预测水平。背景
3.免疫疗法用于治疗多种癌症和自身免疫病症。尽管已知免疫检查点阻断疗法是用于各种恶性肿瘤的有效的癌症治疗类型,但是一致地预测受试者对这些疗法的响应的诊断生物标志物仍然是难以解决的。考虑到免疫系统对免疫疗法的抗性的高度可变和复杂的性质,以及与治疗相关的潜在毒性,精确预测对某些免疫疗法的治疗响应可能是具有挑战性的。
4.免疫基因组学已经作为一种可以确定免疫疗法的治疗功效的技术而出现。这种技术可以导致对癌症的有效治疗的确定,并且可以有助于发现几种新的治疗剂,诊断剂和方法。例如,免疫基因组学可用于鉴定新抗原,而新抗原可有助于精确癌症治疗和诊断的发展。此外,基因组数据如变体调用可以提供对复杂免疫系统应答和对癌症免疫疗法的抗性的深刻理解。然而,使用靶向诊断癌症组的常规技术提供了有限的数据量,这对于开发整合的综合生物标志物可能是不可靠的。简要概述
5.在一些实施方案中,提供了用于确定综合生物标志物得分的方法和系统,所述综合生物标志物得分鉴定受试者对特定类型的免疫疗法治疗的预测水平的响应性。免疫基因组学分析系统访问通过处理受试者的生物样品产生的基因组数据和转录物组数据。在一些情况下,生物样品包括一种或多种癌细胞。基因组数据可鉴定生物样品中的一条或多条dna序列,其中可进行全外显子组测序以鉴定一条或多条dna序列。转录物组数据可鉴定生物样品中的一条或多条rna序列,其中转录物组测序可用于鉴定一条或多条rna序列。另外地或可选地,基因组和转录物组数据可以从包括受试者的生物样品和参考生物样品的样品对产生,其中参考生物样品不包括一种或多种癌细胞。
6.免疫基因组学分析系统处理基因组数据以产生一组基因组度量。所述一组基因组度量中的每一个可以代表对应于一条或多条dna序列的相应dna序列的一个或多个特征。在一些情况下,所述一组基因组度量包括:(i)定量或分类度量,其代表一条或多条dna序列中的一个或多个体细胞突变中的每一个的一个或多个特征;(ii)分类度量,其指示在所述生物样品的至少一种人类白细胞抗原(hla)基因中是否已经发生杂合性丢失;和(iii)代表预测的肿瘤突变负荷的定量或分类度量。关于hla的杂合性丢失,可以通过将基因组数据应用
于hla缺失-鉴定机器学习模型来产生相应的分类度量。
7.免疫基因组学分析系统处理转录物组数据以产生一组转录物组度量。所述一组转录物组度量中的每一个可代表对应于一组肽的一个或多个特征,所述一组肽由一条或多条rna序列的相应rna序列翻译而成。在一些情况下,所述一组转录物组度量包括:(i)定量或分类度量,其代表生物样品的预测的新抗原负荷;(ii)定量或分类度量,其代表从所述生物样品检测到的一种或多种候选新抗原中的每一种的一个或多个特征;(iii)定量或分类度量,其代表检测到细胞表面呈递丢失的一种或多种hla蛋白中的每一种的一个或多个特征;(iv)定量或分类度量,其代表对应于hla基因的一个或多个特征,所述hla基因编码检测到细胞表面呈递丢失的一种或多种hla蛋白;(v)定量或分类度量,其代表对应于免疫细胞的序列的表达水平;和(vi)定量或分类度量,其代表从所述生物样品检测到的一种或多种t细胞受体的表达水平。对于检测到细胞表面呈递丢失的hla蛋白,可以通过将基因组和转录物组数据应用于新抗原呈递预测机器学习模型来产生相应的度量。
8.免疫基因组学分析系统产生从一组基因组度量和一组转录物组度量得到的综合生物标志物得分,并基于综合生物标志物得分确定受试者对特定类型的免疫疗法治疗的响应性的预测水平。在一些情况下,免疫基因组学分析系统通过以下步骤生成综合生物标志物得分:(i)将一组基因组度量中的每个基因组度量与基于一组转录物组度量中相应的转录物组度量确定的权重值进行加权;和(ii)使用加权的基因组度量生成综合生物标志物得分。
9.免疫基因组学分析系统输出对应于受试者的响应性的预测水平的结果。结果可以是基于受试者对特定治疗的响应性的预测水平来鉴定以下的报告:(i)特定治疗的治疗建议;(ii)向人类受试者施用特定治疗的建议;和/或(iii)不向人类受试者施用特定治疗的建议。在一些实施方案中,将推荐的治疗施用于人类受试者。
10.在一些实施方案中,提供了一种计算机程序产品,其有形地体现在非暂时性机器可读存储介质中,并且包括被配置成使一个或多个数据处理器执行本文所公开的一个或多个方法的一部分或全部的指令。
11.本公开的一些实施方案包括系统,其包括一个或多个数据处理器。在一些实施方案中,所述系统包括包含指令的非暂时性计算机可读存储介质,当所述指令在所述一个或多个数据处理器上执行时,使得所述一个或多个数据处理器执行本文所公开的一个或多个方法的一部分或全部和/或一个或多个过程的一部分或全部。本公开的一些实施方案包括有形地体现在非暂时性机器可读存储介质中的计算机程序产品,所述计算机程序产品包括被配置成使一个或多个数据处理器执行本文所公开的一个或多个方法的一部分或全部和/或一个或多个过程的一部分或全部的指令。
12.所使用的术语和表述被用作描述性术语而不是限制性术语,并且在使用这样的术语和表述时不打算排除所示出和描述的特征或其部分的任何等同物,但是应当认识到,在所要求保护的本发明的范围内,各种修改是可能的。因此,应当理解,尽管已经通过实施方案和可选特征具体公开了所要求保护的本发明,但是本领域的技术人员可以对本文公开的概念进行修改和改变,并且这些修改和改变被认为在由所附权利要求限定的本发明的范围内。附图的简要说明
13.当参考以下附图阅读以下详细描述时,将更好地理解本公开的特征,实施方案和优点。本专利或申请文件包含至少一幅彩色的附图。本专利或专利申请公开的带有彩色附图的副本将由专利局在请求和支付必要的费用时提供。
14.图1显示了根据一些实施方案的用于从生物样品产生基因组数据和转录物组数据的示意图的实例。
15.图2a-b显示了对应于临床群组受试者的基因组和转录物组数据中的致癌变化的统计数据。
16.图3a-c显示了与鉴定与免疫系统应答相关的差异表达基因的转录物组度量相对应的统计数据。
17.图4显示了对应于每个差异调节的免疫途径的归一化的富集得分的统计数据。
18.图5a-c显示了与鉴定t细胞受体表达水平的转录物组度量相对应的统计数据。
19.图6显示了鉴定第一组响应受试者和第二组非响应受试者之间的富集得分的比较的一组框图。
20.图7a-b显示了与鉴定跨各种基因和疾病位置的新抗原负荷的转录物组度量相对应的统计数据。
21.图8a-f显示了鉴定跨各个受试者的新抗原负荷得分的统计数据,其中新抗原负荷得分可以预测用免疫疗法治疗的受试者的响应性。
22.图9a-f显示了鉴定与发现群组的每个受试者样品中存在的突变相关的一个或多个特征的统计数据。
23.图10显示了鉴定跨各种驱动突变、疾病位置和受试者组的肿瘤突变负荷的多组框图。
24.图11a-d显示了鉴定跨各个受试者的综合生物标志物得分的统计数据,其中综合生物标志物得分表明在预测用免疫疗法治疗的受试者的响应性方面的表现改善。
25.图12a-b显示了鉴定跨各个受试者的综合生物标志物得分的统计数据,其中综合生物标志物得分表明在预测群组中受试者的无进展和总体生存率方面的表现改善。
26.图13a-b显示了鉴定hla基因的体细胞突变的统计数据,所述hla基因的体细胞突变可能有助于降低新抗原呈递的概率。
27.图14a-b显示了鉴定特定受试者的正常样品和相应肿瘤样品之间的hla序列的比较的一组图的实例。
28.图15包括说明根据一些实施方案产生综合生物标志物得分的方法的实例的流程图。详细描述i.概述
29.如上所述,检查点抑制剂疗法的功效可取决于各种生物因素,包括肿瘤,相应的肿瘤微环境和相应的免疫系统之间的复杂相互作用。已经讨论了用于鉴定免疫系统对免疫疗法的应答的多种生物标志物,包括pd-l1表达,基于干扰素(ifn)-γ的标签,肿瘤突变负荷,错配修复缺陷,遗传改变(包括抗原呈递机制内的那些),hla杂合性丢失和t细胞组库多样性。
30.如可影响免疫系统对免疫检查点阻断疗法的应答的各种生物因子所示,已经有越
来越多的关于可掺入各种生物因子并准确预测免疫系统对免疫疗法的应答的整合生物标志物的尝试。例如,常规技术已经将与样品的免疫原性和新抗原克隆结构相对应的信息组合,以预测免疫系统对免疫检查点阻断的应答。通过这些常规技术产生的结果已经尝试确定患有黑色素瘤,肺癌和肾癌的受试者的预后。尽管这些常规技术已经产生了稍微积极的结果,但是常规技术在产生能够一致且准确地预测免疫系统应答的数据方面仍然存在不足。这种挑战可以归因于驱动对肿瘤的免疫应答的复杂机制。此外,这些常规技术需要来自受试者的大量样品,其在一些情况下(例如受试者的年龄,受试者是怀孕的)可能是侵入性的并且难以获得。
31.为了解决常规系统的至少上述缺陷,本技术的技术可用于确定综合生物标志物得分,所述得分鉴定受试者对特定类型的免疫疗法治疗的的响应性的预测水平。免疫基因组学分析系统访问通过处理受试者的生物样品产生的基因组数据和转录物组数据。在一些情况下,生物样品包括一种或多种癌细胞。基因组数据可鉴定生物样品中的一条或多条dna序列,其中可进行全外显子组测序以鉴定一条或多条dna序列。转录物组数据可鉴定生物样品中的一条或多条rna序列,其中转录物组测序可用于鉴定一条或多条rna序列。另外地或可选地,基因组和转录物组数据可以从包括受试者的生物样品和参考生物样品的样品对产生,其中参考生物样品不包括一种或多种癌细胞。
32.免疫基因组学分析系统处理基因组数据以产生一组基因组度量。一组基因组度量中的每一个可以代表对应于一条或多条dna序列的相应dna序列的一个或多个特征。在一些情况下,所述一组基因组度量包括:(i)定量或分类度量,其代表一条或多条dna序列中的一个或多个体细胞突变中的每一个的一个或多个特征;(ii)分类度量,其指示在所述生物样品的至少一种人类白细胞抗原(hla)基因中是否已经发生杂合性丢失;和(iii)代表预测的肿瘤突变负荷的定量或分类度量。关于hla的杂合性丢失,可以通过将基因组数据应用于hla缺失-鉴定机器学习模型来产生相应的分类度量。
33.免疫基因组学分析系统处理转录物组数据以产生一组转录物组度量。所述一组转录物组度量中的每一个可代表对应于一组肽的一个或多个特征,所述一组肽由一条或多条rna序列的相应rna序列翻译而成。在一些情况下,所述一组转录物组度量包括:(i)定量或分类度量,其代表生物样品的预测的新抗原负荷;(ii)定量或分类度量,其代表从所述生物样品检测到的一种或多种候选新抗原中的每一种的一个或多个特征;(iii)定量或分类度量,其代表检测到细胞表面呈递丢失的一种或多种hla蛋白中的每一种的一个或多个特征;(iv)定量或分类度量,其代表对应于hla基因的一个或多个特征,所述hla基因编码检测到细胞表面呈递丢失的一种或多种hla蛋白;(v)定量或分类度量,其代表对应于免疫细胞的序列的表达水平;和(vi)定量或分类度量,其代表从所述生物样品检测到的一种或多种t细胞受体的表达水平。对于检测到细胞表面呈递丢失的hla蛋白,可以通过将基因组和转录物组数据应用于新抗原呈递预测机器学习模型来产生相应的度量。
34.免疫基因组学分析系统产生从一组基因组度量和一组转录物组度量得到的综合生物标志物得分,并基于综合生物标志物得分确定受试者对特定类型的免疫疗法治疗的响应性的预测水平。在一些情况下,免疫基因组学分析系统通过以下步骤产生综合生物标志物得分:(i)将一组基因组度量中的每个基因组度量与基于一组转录物组度量中相应的转录物组度量确定的权重值进行加权;和(ii)使用加权的基因组度量生成综合生物标志物得
分。
35.免疫基因组学分析系统输出对应于受试者的预测的响应性水平的结果。结果可以是基于受试者对特定治疗的响应性的预测水平来鉴定以下的报告:(i)特定治疗的治疗建议;(ii)向人类受试者施用特定治疗的建议;和/或(iii)不向人类受试者施用特定治疗的建议。在一些实施方案中,将推荐的治疗施用于人类受试者。
36.因此,本公开的实施方案通过基于经验证的,增强的基于外显子组和基于转录物组的肿瘤剖析平台来产生综合生物标志物得分,从而提供优于常规技术的技术优势。特别地,综合生物标志物得分可以从代表各种肿瘤和免疫相关分子机制的特征的度量确定,同时最小化用于产生度量的生物样品的量。这种技术可以提高相应受试者的诊断,预后和/或治疗建议的精确性,而不需要获得大量生物样品的侵入性程序。因此,本公开的实施方案提供了一种综合免疫基因组学框架,用于通过鉴定驱动对免疫疗法的响应和抗性的生物机制来精确地预测对免疫疗法的响应。
37.尽管本文已经示出和描述了本公开的发明的各个实施方案,但是对于本领域技术人员显而易见的是,这些实施方案仅通过示例的方式来提供。在不脱离本发明的情况下,本领域技术人员可以进行多种变化,改变和替换。应当理解,在实践本文所述的任何一项发明时,可以采用本文所述的本发明的实施方案的各种替代方案。ii.定义
38.虽然在本文已经示出和描述了本发明的各个实施方案,但是对于本领域技术人员显而易见的是,这些实施方案仅通过示例的方式来提供。在不脱离本发明的情况下,本领域技术人员可以进行多种变化,改变和替换。应当理解,可以采用本文所述的本发明的实施方案的各种替代方案。
39.如本文所用,术语“癌症”或“恶性肿瘤”通常是指身体细胞不停的分裂并扩散到周围组织中的相关疾病的集合。癌症可以几乎在体内的任何地方开始,并且当去除和替换老的、异常的或受损的细胞的有序过程被破坏时出现,并且当这些细胞应该死亡时,它们存活,或者不需要它们时仍形成新的细胞。这些细胞不停地分裂并且能够扩散到附近和远离它们的原点的组织中并侵入它们。
40.如本文所用,术语“新抗原”通常是指先前未被免疫系统识别的新形成的抗原。新抗原可以由由于肿瘤突变而形成的改变的肿瘤蛋白产生。新抗原可构成可加载到mhc i类和ii类分子上并呈递给t细胞的体细胞突变的子集。这些新抗原可被免疫系统视为内源性肿瘤特异性(非自身)靶标。
41.如本文所用,术语“肿瘤微环境”(tumor microenvironment)是指肿瘤周围的环境,包括周围的血管、免疫细胞、成纤维细胞、信号传导分子和细胞外基质。肿瘤及其微环境密切相关并总是呈动态互惠式相互作用。肿瘤进展受癌细胞与其环境的影响并塑造治疗响应和抗性。
42.如本文所用,术语“生物标志物”是指代谢物或由其衍生的小分子,其与来自具有第二表型(例如,不具有疾病)的受试者或受试者组的生物样品相比,差异性地存在于(即,增加或减少)来自具有第一表型(例如,具有疾病)的受试者或受试者组的生物样品中。生物标志物可以以任何水平差异性地存在,但通常以增加至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、
至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%、至少100%、至少110%、至少120%、至少130%、至少140%、至少150%或更多的水平存在;或者通常以降低至少5%、至少10%、至少15%、至少20%、至少25%、至少30%、至少35%、至少40%、至少45%、至少50%、至少55%、至少60%、至少65%、至少70%、至少75%、至少80%、至少85%、至少90%、至少95%或100%(即,不存在)的水平存在。生物标志物优选以统计学显著的水平差异性地存在。
43.如本文所用,术语“水平”是指一种或多种生物标志物的水平,其意指样品中生物标志物的绝对或相对量或浓度。
44.如本文所用,术语“参考谱”是指指示健康受试者或疾病状态、病况或身体病症中的一种或多种的代谢谱。在参考谱内,将存在一种或多种生物标志物(代谢物或由其衍生的小分子)的参考水平,所述参考水平可以是所述一种或多种生物标志物的绝对或相对量或浓度、所述一种或多种生物标志物的存在或不存在、所述一种或多种生物标志物的量或浓度范围、所述一种或多种生物标志物的最小和/或最大量或浓度、所述一种或多种生物标志物的平均量或浓度和/或所述一种或多种生物标志物的中值量或浓度。
45.如本文所用,术语“统计学上显著的”是指,如使用参数或非参数统计所确定的,例如但不限于anova或wilcoxon秩和检验,至少约95%的置信度水平,优选至少约97%的置信度水平,更优选至少约98%的置信度水平,最优选至少约99%的置信度水平,其中对于至少约95%的置信度水平,后者表示为p《0.05。本文所用的术语“免疫检查点阻断”通常是指通过抑制免疫抑制分子从而防止免疫应答终止或使t淋巴细胞在免疫应答期间耗尽而集中于免疫应答终止的疗法。
46.每当术语“至少”、“大于”或“大于或等于”在两个或更多个数值的序列中的第一数值之前时,术语“至少”、“大于”或“大于或等于”适用于该数值序列中的每一个数值。例如,大于或等于1、2或3等同于大于或等于1、大于或等于2、或大于或等于3。
47.每当术语“不超过”、“小于”或“小于或等于”在两个或更多个数值的序列中的第一数值之前时,术语“不超过”、“小于”或“小于或等于”适用于该数值序列中的每一个数值。例如,小于或等于3、2或1等同于小于或等于3、小于或等于2、或小于或等于1。
48.当与权利要求和/或说明书中的术语“包含”结合使用时,词语“一个”或“一种”的使用可以意味着“一个”,但是它也与“一个或多个”、“至少一个”和“一个或多于一个”的含义一致。
49.权利要求中使用的术语“或”用于表示“和/或”,除非明确地仅指代替代方案或替代方案是互斥的,尽管本公开支持仅指代替代方案的定义和“和/或”。本文所用的“另一种”可以是指至少第二种或更多种。
50.术语“包含(comprise)”、“具有(have)”和“包括(include)”是开放式连接动词。这些动词中的一个或多个的任何形式或时态,例如“包含(comprises)”、“包含(comprising)”、“具有(has)”、“具有(having)”、“包括(includes)”和“包括(including)”也是开放式的。例如,“包含”、“具有”或“包括”一个或多个步骤的任何方法不限于仅具有那些一个或多个步骤,并且还涵盖其他未列出的步骤。iii.免疫疗法治疗和免疫系统应答机制a.肿瘤微环境
51.免疫系统可检测多种抗原,例如病毒,寄生虫或变应原,癌症,并在体内引发对抗外来物质,异常细胞和/或组织的应答。癌性生长,包括恶性癌性生长,也可以被受试者的免疫细胞识别并触发免疫应答。免疫细胞的活化可触发许多细胞内信号传导途径,其需要严格控制以实现适当的免疫应答。癌性生长可以与其微环境密切相互作用。肿瘤不仅可以由异质的癌细胞群组成,而且可以由多种驻留和浸润的宿主细胞,分泌的因子和细胞外基质蛋白组成。癌症和肿瘤进展可以受到癌细胞与这种肿瘤微环境的相互作用的深远影响,这可以最终决定肿瘤根除,转移,治疗响应或抗性。肿瘤微环境对癌症进展的机制可以在靶向肿瘤微环境的组分方面,例如在免疫检查点抑制剂疗法中提供治疗途径。
52.肿瘤微环境,特别是实体瘤中的肿瘤微环境可能对免疫细胞如效应t细胞保持敌对。免疫抑制信号的筑起和肿瘤微环境内必需营养物的缺乏可能导致t细胞耗尽。克服肿瘤微环境并确定对治疗的早期预测响应可能是促进免疫疗法在根除肿瘤的癌细胞中的效率的重要因素。癌细胞的代谢重编程和可塑性以适应它们的快速增殖可能是恶性癌症中治疗抗性的重要机制。几种免疫细胞类型存在于肿瘤微环境中并且可能在癌症进展中具有积极作用,包括但不限于巨噬细胞、b细胞、t细胞、嗜中性粒细胞和树突细胞。b.肿瘤逃逸机制
53.从赘生性起始进展到恶性可能部分地由于免疫监视的失败而发生。癌细胞可逃避免疫识别和消除并产生免疫抑制性微环境。由于癌细胞的高消耗,区域中的天然免疫细胞可能面对营养物缺乏的环境。癌细胞代谢的多种代谢副产物如乳酸盐和糖酵解的终产物可能对天然免疫细胞有害,损害它们的分化、活化、适合性、抗肿瘤功能,并使它们广泛地不能与癌细胞竞争。
54.肿瘤微环境中的代谢变化如缺氧也可能影响髓样细胞的分化程序,从而改变它们的抗原呈递特性。缺氧介导的表达可选择性地上调抑制配体的表达,从而促进t细胞免疫抑制。当肿瘤微环境中的癌症介导的代谢变化影响免疫微环境的细胞组成和功能时,靶向癌细胞的代谢变化可以影响癌细胞的生长和进展,并且通过改变免疫细胞的代谢程序和它们的抗肿瘤功能提供用于改善抗肿瘤免疫的治疗靶标。c.免疫疗法
55.代谢过程可以在静止条件下以及在致病过程如感染,炎症,癌症和自身免疫中调节免疫细胞应答。在这些复杂的病况中,免疫疗法可以提供新的治疗途径。巨噬细胞以及其它免疫细胞显示出依赖于疾病病理的代谢可塑性。肿瘤浸润性淋巴细胞可以是肿瘤微环境的显著部分,并且与改善的预后和对疗法的响应相关(cogdill,andrews和wargo 2017tomioka等人,2018)。
56.免疫疗法可激活受试者的免疫系统以对抗癌症。为了用免疫疗法有效根除癌细胞,t细胞或其它免疫细胞可以识别由人类白细胞抗原(hla)呈递的肿瘤肽。hla或主要组织相容性复合物可以是参与抗原呈递的蛋白质,并且可以由hla基因编码。检查点抑制剂疗法已经显示出有意义的抗肿瘤活性,受试者反应受多种生物因素的影响,包括肿瘤、肿瘤微环境和免疫系统之间的复杂相互作用(hodi等人2010;larkin,ho和wolchok 2015hugo等人2016;ribas等人2016;wolchok等人2017)。
57.免疫检查点阻断疗法可用于促进或抑制t细胞活化。免疫应答可以包括起始阶段和活化阶段,其中免疫系统识别危险信号并被先天信号活化以对抗危险。该反应可以是抗
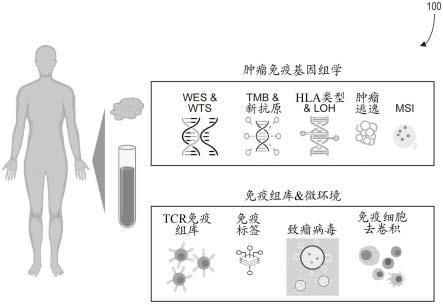
感染和癌症的第一步之一,但是一旦控制了危险,就需要关闭,因为该活化的持续可能引起组织损伤。在免疫系统活化后,随后是终止阶段,其中内源性免疫抑制分子可以阻止免疫应答以防止损伤。在癌症免疫疗法中,治疗方法典型地增强免疫应答的起始和活化以增加t-淋巴细胞对抗癌症的出现和功效。免疫检查点阻断疗法可集中于终止免疫应答,其通过抑制免疫抑制分子,从而防止免疫应答终止或唤醒在免疫应答期间变得耗尽的t-淋巴细胞。阻断负调节免疫检查点可恢复耗尽的免疫细胞杀死它们浸润的癌症的能力,并驱动存活的癌细胞进入休眠状态。
58.免疫检查点可以是免疫系统固有的共刺激和抑制元件。免疫检查点可以帮助维持自身耐受性和调节生理免疫应答的持续时间和幅度,以防止当免疫系统应答致病性感染时对组织的损伤。当t细胞识别肿瘤细胞特征性抗原时,也可以引发免疫应答。共刺激和抑制信号之间的平衡可用于控制来自t细胞的免疫应答,其可由免疫检查点蛋白调节。在t细胞在胸腺中成熟和活化后,t细胞可行进至炎症和损伤部位以进行修复功能。t细胞功能可以通过直接作用或通过参与免疫系统的细胞因子和膜配体的募集而发生。涉及t细胞成熟、活化、增殖和功能的步骤可以通过共刺激和抑制信号,即通过免疫检查点蛋白来调节。肿瘤可以异常调节检查点蛋白功能作为免疫抵抗机制。因此,对检查点蛋白调节剂的开发可具有治疗价值。免疫检查点分子的非限制性实例包括ctla4和pd-1。这些检查点分子可以在途径中操纵il-2的上游。iv.用于预测免疫系统对免疫疗法的应答的生物标志物的实例
59.免疫检查点分子可以是免疫球蛋白超家族的成员并且可以是防止不受控制的免疫应答的抑制性受体。适应性免疫应答可以由这些检查点分子控制,所述检查点分子可以用于维持自身耐受性和最小化在免疫应答期间可能发生的旁系组织损伤。已经提出了许多对免疫检查点阻断应答的生物标志物,包括pd-l1表达、基于干扰素(ifnγ)的标签、肿瘤突变负荷、微卫星不稳定性(msi)和错配修复缺陷、遗传改变,包括抗原呈递机制内的那些遗传改变、hla的杂合性丢失以及t细胞组库多样性(herbst等人2014;gao等人2016;zaretsky等人2016;roh等人2017sade-feldman等人2017;mariathasan等人2018;chowell等人2019)。
60.由于可影响对免疫检查点阻断疗法的响应的生物特征的多样性,已经有越来越多的对鉴定整合了多种生物特征以更好地预测对免疫疗法的响应的生物标志物的尝试(charoentong等人,2017)。在一项这样的尝试中,将纯度校正的肿瘤突变负荷与受体酪氨酸激酶(rtk)突变、hla突变和吸烟标签相结合的标签被用于预测非小细胞肺癌(nsclc)中的免疫检查点阻断响应(anagnostou等人2020),而黑色素瘤研究组合了基因组、转录物组和临床数据以预测对免疫检查点阻断的响应(liu等人2019)。
61.新抗原可以构成体细胞突变的子集,其可以装载到mhc i类和ii类分子上并被呈递给t细胞。这些新抗原可被免疫系统视为内源性肿瘤特异性(非自身)靶标。免疫检查点阻断被认为是利用细胞毒性(cd8+)t细胞检测和破坏在其h-ic i类分子上显示新抗原的癌细胞的能力(schumacher和schreiber 2015)。整合了免疫原性和新抗原克隆结构的工作在患有黑色素瘤、肺癌和肾癌的受试者中预测了对免疫检查点阻断的响应和预后,表明了生物标志物的广泛适用性(lu等人2020)。
62.最近,在使用基因表达分析,代谢组学和蛋白质组学方法鉴定用于癌症诊断和进
展的替代生物标志物方面的努力增加。基因表达分析可提供对杂合性丢失的深刻理解,杂合性丢失是可导致整个基因和周围染色体区域丢失的交叉染色体事件。杂合性丢失可指示在癌症的丢失区域中不存在功能性肿瘤抑制基因。肿瘤抑制基因可以通过这种丢失或通过点突变而失活,所述点突变不会使肿瘤抑制基因保护身体免受癌性生长。hla的杂合性丢失检测可以是泛癌生物标志物。v.用于产生综合生物标志物得分的技术
63.如本文所述,由免疫基因组学分析系统产生的综合生物标志物得分可以将与抗原呈递机制中的损伤事件(例如,hla的杂合性丢失)有关的信息与预测的新抗原结合,以对受试者对免疫疗法的响应进行分级。综合生物标志物得分优于常规的单一分析物生物标志物,表明捕获肿瘤逃逸的多个方面的复杂模型可以提供受试者方向的更强健的分级。此外,这种数据密集型生物标志物在临床上是实用的,使用有限的肿瘤组织在各种临床群组中实现了全面的肿瘤剖析。这些发现提供了在晚期癌症受试者中响应的精确的综合生物标志物,以及支持在临床环境中使用全外显子组和转录物组数据的证据。a.生成基因组和转录物组数据1.生物样品
64.图1显示了根据一些实施方案的用于从生物样品产生基因组数据和转录物组数据的示意图100的实例。例如,示意图100包括从受试者选择生物样品,其中生物样品包括癌细胞。在一些情况下,从受试者收集治疗前血液正常样品和肿瘤样品。例如,可以从接受了抗pd-1疗法的具有不可切除的iii/iv期黑色素瘤的受试者收集治疗前血液正常样品和肿瘤样品。
65.可以处理生物样品以产生受试者的免疫基因组学谱,其中所述谱可包括全面的肿瘤突变信息,基因表达定量,新抗原表征,hla(分型,突变和杂合性丢失),t细胞受体组库剖析,微卫星不稳定性检测,肿瘤病毒鉴定和肿瘤微环境剖析。然后可以将谱数据与临床结果,和针对受试者计算的综合生物标志物得分一起分析,以便鉴定对特定免疫疗法治疗的响应性的预测水平。
66.样品可取自受试者。样品可以从血液(例如,全血),血浆,血清,脐带血,绒毛膜,羊水,灌洗液(例如,支气管肺泡,胃,腹部,导管,耳,关节镜),活检样品(例如,来自植入前胚胎),穿刺(celocentesis)样品,胎儿有核细胞或胎儿细胞残留物,胆汁,母乳,尿液,唾液,粘膜分泌物,痰,粪便,汗液,阴道流体,来自积液的流体(例如睾丸的积液),阴道冲洗液,胸腔流体,腹水,脑脊液,支气管肺泡灌洗液,来自乳头的排出流体,来自身体的不同部分(例如,甲状腺、乳房)的抽吸流体,泪液,、胚胎细胞或胎儿细胞(例如,胎盘细胞)。在一些实施方案中,通过扎脚跟或手指,从头皮静脉或通过耳垂穿刺获得血液样品。生物样品可以是流体或组织样品(例如皮肤样品)。生物样品可以包括任何组织或来源于活体或死亡受试者的材料。生物样品可以是无细胞样品。生物样品可以包含蛋白质或核酸(例如dna或rna或其片段)。样品可以是固定的或者可以不是固定的。样品可以被包埋或者可以是自由的。样品可以是福尔马林固定的石蜡包埋的样品。
67.生物样品可以包括一种或多种核酸分子。核酸分子可以是dna分子,rna分子(例如mrna、crna或mirna)和dna/rna杂合体。dna分子的实例包括但不限于双链dna,单链dna,单链dna发夹,cdna,基因组dna。核酸可以是rna分子,例如双链rna,单链rna,ncrna,rna发夹
和mrna。ncrna的实例包括但不限于sirna、mirna、snorna、pirna、tirna、pasr、tasr、atasr、tssa-rna、snrna、re-rna、uarna、x-ncrna、hy rna、usrna、snar和vtrna。2.测序
68.为了从生物样品产生对应于基因组数据的dna序列,可以进行全外显子组文库的制备和测序。从生物样品中提取dna,处理,并进行全外显子组测序。可以使用来自肿瘤和正常血液样品的dna构建全外显组子捕获文库。在一些情况下,靶探针用于增强生物医学和临床相关基因的覆盖。可修改方案以产生约250bp的平均文库插入长度。对测序读数进行质量控制处理(例如,经由fastqc)以提供fastq文件。将fastq文件与参考基因组比对以产生bam文件。
69.为了从生物样品产生对应于转录物组数据的rna序列,可以进行转录物组测序。在一些情况下,转录物组测序包括微阵列和rna-seq。微阵列可经配置以经由其与互补探针阵列的杂交来测量确定的一组转录物的丰度。rna-seq可以指对生物样品中转录物的互补dna进行测序,其中互补dna的丰度来自每种转录物的计数数目。
70.在一些情况下,样品处理包括核酸样品处理和随后的核酸样品测序。核酸样品中的一些或全部可以被测序以提供序列信息,其可以被存储或以其它方式维持在电子,磁或光存储位置。可以借助于计算机处理器来分析序列信息,并且可以将所分析的序列信息存储在电子存储位置中。电子存储位置可以包括从核酸样品产生的序列信息和分析的序列信息的库或集合。
71.一些实施方案可以包括使用全基因组测序。在一些情况下,全基因组测序用于鉴定人体中的变体。在一些情况下,测序可包括对基因组的一部分进行深度测序。例如,基因组的一部分可以是至少约50;75;100;125;150;175;200;225;250;275;300;350;400;450;500;550;600;650;700;750;800;850;900;950;1,000;1100;1200;1300;1400;1500;1600;1700;1800;1900;2,000;3,000;4,000;5,000;6,000;7,000;8,000;9,000;10,000;15,000;20,000;30,000;40,000;50,000;60,000;70,000;80,000;90,000;100,000或更多个碱基或碱基对。在一些情况下,基因组可以被测序超过1百万,2百万,3百万,4百万,5百万,6百万,7百万,8百万,9百万,1千万或超过1千万个碱基或碱基对。在一些情况下,基因组可以在整个外显子组上测序(例如,全外显子组测序)。在一些情况下,深度测序可包括获得对基因组的一部分的多个读数。例如,获得多个读数可以包括在基因组的一部分上的至少2、3、4、5、6、7、8、9、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900、1000、10,000个读数或超过10,000个读数。
72.一些实施方案可以包括通过深度测序检测低等位基因部分。在一些情况下,通过下一代测序进行深度测序。在一些情况下,通过避免易错区域来完成深度排序。在一些情况下,易错区域可包括近序列重复的区域,异常高或低%gc的区域,近均聚物的区域,二核苷酸和三核苷酸区域以及近其它短重复的区域。在一些情况下,易错区域可包括导致dna测序错误(例如,均聚物序列中的聚合酶滑移)的区域。
73.一些实施方案可以包括在样品中的一种或多种核酸分子上进行一个或多个测序反应。一些实施方案可以包括在样品中的一种或多种核酸分子上进行1个或更多个,2个或更多个,3个或更多个,4个或更多个,5个或更多个,6个或更多个,7个或更多个,8个或更多个,9个或更多个,10个或更多个,15个或更多个,20个或更多个,30个或更多个,40个或更多
个,50个或更多个,60个或更多个,70个或更多个,80个或更多个,90个或更多个,100个或更多个,200个或更多个,300个或更多个,400个或更多个,500个或更多个,600个或更多个,700个或更多个,800个或更多个,900个或更多个,或1000个或更多个测序反应。测序反应可以同时,顺序或其组合进行。测序反应可包括全基因组测序或外显子组测序。测序反应可以包括maxim-gilbert,链终止或高通量系统。可选地或另外地,测序反应可包括helicoscopetm单分子测序,纳米孔dna测序,lynx therapeutics大规模平行标签测序(mpss),454焦磷酸测序,单分子实时(rnap)测序,illumina(solexa)测序,solid测序,ion torrenttm,离子半导体测序,单分子smrt(tm)测序,多克隆测序,dna纳米球测序,visigen生物技术方法或以上的组合。可选地或另外地,测序反应可包括一个或多个测序平台,包括但不限于由illumina提供的genome analyzer iix,hiseq和miseq,单分子实时(smrttm)技术,如由pacific biosciences(california)和solexa测序仪提供的pacbio rs系统,真单分子测序(tsmstm)技术,例如helicos inc.提供的heliscopetm测序仪(cambridge,ma)。测序反应也可以包括电子显微镜或化学敏感场效应晶体管(chemfet)阵列。在一些方面,测序反应包括毛细管测序,下一代测序,sanger测序,通过合成测序,通过连接测序,通过杂交测序,单分子测序或以上的组合。通过合成测序可以包括可逆终止子测序,进行性单分子测序,顺序流测序或以上的组合。顺序流测序可包括焦磷酸测序,ph介导的测序,半导体测序或以上的组合。
74.一些实施方案可以包括进行至少一个长读测序反应和至少一个短读测序反应。长读测序反应和/或短读测序反应可以在核酸分子子集的至少一部分上进行。长读测序反应和/或短读测序反应可以在核酸分子的两个或更多个子集的至少一部分上进行。长读测序反应和短读测序反应都可以在核酸分子的一个或多个子集的至少一部分上进行。
75.对一种或多种核酸分子或其子集的测序可以包括至少约5;10;15;20;25;30;35;40;45;50;60;70;80;90;100;200;300;400;500;600;700;800;900;1,000;1500;2,000;2500;3,000;3500;4,000;4500;5,000;5500;6,000;6500;7,000;7500;8,000;8500;9,000;10,000;25,000;50,000;75,000;100,000;250,000;500,000;750,000;10,000,000;25,000,000;50,000,000;100,000,000;250,000,000;500,000,000;750,000,000;1,000,000,000个或更多个测序读数。
76.测序反应可包括对一种或多种核酸分子的至少约50;60;70;80;90;100;110;120;130;140;150;160;170;180;190;200;210;220;230;240;250;260;270;280;290;300;325;350;375;400;425;450;475;500;600;700;800;900;1,000;1500;2,000;2500;3,000;3500;4,000;4500;5,000;5500;6,000;6500;7,000;7500;8,000;8500;9,000;10,000;20,000;30,000;40,000;50,000;60,000;70,000;80,000;90,000;100,000个或更多个碱基或碱基对进行测序。测序反应可包括对一种或多种核酸分子的至少50;60;70;80;90;100;110;120;130;140;150;160;170;180;190;200;210;220;230;240;250;260;270;280;290;300;325;350;375;400;425;450;475;500;600;700;800;900;1,000;1500;2,000;2500;3,000;3500;4,000;4500;5,000;5500;6,000;6500;7,000;7500;8,000;8500;9,000;10,000;20,000;30,000;40,000;50,000;60,000;70,000;80,000;90,000;100,000个或更多个连续碱基或碱基对进行测序。
77.优选地,在本发明的方法中使用的排序技术产生至少100个读数/运行,至少200个
读数/运行,至少300个读数/运行,至少400个读数/运行,至少500个读数/运行,至少600个读数/运行,至少700个读数/运行,至少800个读数/运行,至少900个读数/运行,至少1000个读数/运行,至少5,000个读数/运行,至少10,000个读数/运行,至少50,000个读数/运行,至少100,000个读数/运行,至少500,000个读数/运行,或至少1,000,000个读数/运行。可选地,在本发明的方法中使用的测序技术产生至少1,500,000个读数/运行,至少2,000,000个读数/运行,至少2,500,000个读数/运行,至少3,000,000个读数/运行,至少3,500,000个读数/运行,至少4,000,000个读数/运行,至少4,500,000个读数/运行,或至少5,000,000个读数/运行。
78.优选地,在本发明的方法中使用的测序技术每个读数可以产生至少约30个碱基对,至少约40个碱基对,至少约50个碱基对,至少约60个碱基对,至少约70个碱基对,至少约80个碱基对,至少约90个碱基对,至少约100个碱基对,至少约110个碱基对,至少约120个碱基对,至少约150个碱基对,至少约200个碱基对,至少约250个碱基对,至少约300个碱基对,至少约350个碱基对,至少约400个碱基对,至少约450个碱基对,至少约500个碱基对,至少约550个碱基对,至少约600个碱基对,至少约700个碱基对,至少约800个碱基对,至少约900个碱基对,或至少约1,000个碱基对。可选地,在本发明的方法中使用的测序技术可以产生长的测序读数。在一些情况下,在本发明的方法中使用的测序技术可以产生至少约1,200个碱基对/读数,至少约1,500个碱基对/读数,至少约1,800个碱基对/读数,至少约2,000个碱基对/读数,至少约2,500个碱基对/读数,至少约3,000个碱基对/读数,至少约3,500个碱基对/读数,至少约4,000个碱基对/读数,至少约4,500个碱基对/读数,至少约5,000个碱基对/读数,至少约6,000个碱基对/读数,至少约7,000个碱基对/读数,至少约8,000个碱基对/读数,至少约9,000个碱基对/读数,至少约10,000个碱基对/读数,20,000个碱基对/读数,30,000个碱基对/读数,40,000个碱基对/读数,50,000个碱基对/读数,60,000个碱基对/读数,70,000个碱基对/读数,80,000个碱基对/读数,90,000个碱基对/读数,或100,000个碱基对/读数。
79.高通量测序系统可以允许在将其掺入生长链之后或当时立即检测测序的核苷酸,即实时或基本上实时地检测序列。在一些情况下,高通量测序每小时产生至少1,000、至少5,000、至少10,000、至少20,000、至少30,000、至少40,000、至少50,000、至少100,000或至少500,000个序列读数;其中每个读数为至少50个,至少60个,至少70个,至少80个,至少90个,至少100个,至少120个,至少150个,至少200个,至少250个,至少300个,至少350个,至少400个,至少450个或至少500个碱基。测序可使用本文所述的核酸如基因组dna,来自rna转录物的cdna或rna作为模板进行。3.比对
80.通过上述测序技术产生的序列读数(例如,dna序列,rna序列)可以被映射到相应的参考基因组(例如,hs37d5参考基因组构建)。在一些情况下,比对流水线执行比对,重复去除和碱基质量得分重新校准以产生基因组和转录物组数据。流水线使用用于重复去除的picard工具包(rrid:scr_006525)和基因组分析工具包(gatk,rrid:scr_001876)来改进序列比对和校正碱基质量得分(bqsr)。然后根据sam(rrid:scr_01095)规范以bam格式返回对齐的序列数据。在一些情况下,基于序列读数与参考基因组的比对来鉴定体细胞变体。
81.在一些情况下,使用star(rrid:scr_015899)比对全转录物组测序,并计算以每百
万转录物(tpm)计的归一化表达值。对于rna测序和比对质量控制,可以鉴定以下度量:平均读数长度,唯一映射的读数的百分比,平均映射读数的对长度,剪接位点的数目,每碱基的错配率,每碱基的缺失/插入率,平均缺失/插入长度,和异常读数对比对,包括染色体间和孤立读数。b.来源于转录物组数据的转录物组度量
82.免疫基因组学分析系统处理对应于生物样品的转录物组数据以产生一组转录物组度量。所述一组转录物组度量中的每一个可代表对应于一组肽的一个或多个特征,所述肽由一条或多条rna序列的相应rna序列翻译而成。在一些情况下,所述一组转录物组度量包括:(i)定量或分类度量,其代表生物样品的预测的新抗原负荷;(ii)定量或分类度量,其代表从所述生物样品检测到的一种或多种候选新抗原中的每一种的一个或多个特征;(iii)定量或分类度量,其代表检测到细胞表面呈递丢失的一种或多种hla蛋白中的每一种的一个或多个特征;(iv)定量或分类度量,其代表对应于hla基因的一个或多个特征,所述hla基因编码检测到细胞表面呈递丢失的一种或多种hla蛋白;和(v)定量或分类度量,其代表从所述生物样品检测到的一种或多种t细胞受体的表达水平。对于检测到细胞表面呈递丢失的hla蛋白,可以通过将基因组和转录物组数据应用于新抗原呈递预测机器学习模型来产生相应的度量。1.免疫浸润标签
83.所述一组转录物组度量可包括代表对应于免疫细胞的序列的表达水平的定量或分类度量。在一些情况下,定量或分类度量是免疫浸润得分,其基于不同类型的肿瘤浸润免疫细胞的量而得到。免疫浸润得分可以使用转录物组数据来计算。例如,可以在单个样品中计算代表基因集合富集的半定量得分。在一些情况下,代表17种细胞类型的一组参考基因表达标签用于产生免疫浸润得分,其中细胞类型可包括恶性细胞,caf,内皮细胞,nk细胞,b细胞,巨噬细胞和cd8
+
和cd4
+
t细胞。
84.为了产生免疫浸润得分,可以使用基因集合富集分析来计算富集得分,当特异于特定细胞类型的基因在感兴趣的样品中靠前地高度表达(即,细胞类型富集在样品中)时,所述富集得分高,而否则则低。相同细胞类型(基因集合)的富集得分可以在样品之间进行比较,剖析受试者的免疫浸润。另外地或可选择地,免疫浸润得分是使用去卷积技术产生的,所述去卷积技术可以定量地估计感兴趣的细胞类型(例如癌细胞)的相对分数。去卷积算法将异源样品的基因表达谱作为不同细胞的基因表达水平的卷积来考虑,并估计在描述细胞类型特异性表达谱的标签矩阵上有影响的未知细胞部分。2.t细胞受体的表达水平
85.所述一组转录物组度量可以包括代表从生物样品中检测到的一种或多种t细胞受体的表达水平的定量的或分类度量。一种或多种t细胞受体的表达水平可以鉴定在生物样品中检测到的克隆淋巴细胞的水平和分布。来自生物样品的淋巴细胞的质量和数量可用于鉴定影响受试者健康和疾病的各种因素。一种或多种t细胞受体的表达水平可以被解释为具有正常的免疫多样性,发育或重建,或者可以被解释为具有炎症,感染,疫苗接种,自身免疫或癌症。在一些情况下,使用多个分析参数来评估生物样品的淋巴样浸润物的质量和数量。分析参数可以包括多样性,丰富度,均匀度,克隆性和熵度量。
86.在一些情况下,一种或多种t细胞受体的表达水平对应于在生物样品中检测到的t
细胞受体β(tcr-β)序列的克隆性。免疫基因组学分析系统处理转录物组数据以剖析tcrβ克隆,其提供tcrβ的增强(相对于标准转录物组约100倍增加)覆盖。可在cdr3序列中筛选出具有移码或过早终止密码子的非生产性克隆,以及具有低于v或j击中阈值的比对得分的低置信度克隆。克隆性然后可以计算为1-pielou的均匀度。3.差异基因表达
87.所述一组转录物组度量可包括代表在转录物组数据中鉴定的每种基因的读数计数的定量度量。例如,每百万序列读数的计数可以通过用生物样品中鉴定的读数总数归一化每种基因的读数计数来计算。在一些情况下,选择特定基因是否应该是定量度量的一部分的阈值。例如,只有在群组的25%或更多的样品中每百万的读取计数》0的基因可被包括用于分析。在一些情况下,使用rlog转化处理剩余的数据并分析差异基因表达。具有调整的p值《0.05和最小log2倍数变化《-0.5或》1的基因被认为是差异表达的。使用各种基因集合,包括但不限于msigdb(molecular signatures database,rrid:scr_016863)标志基因集合和kegg(rrid:scr_012773)基因集合,可以在途径水平上鉴定差异表达基因的生物学意义。4.新抗原呈递预测
88.所述一组转录物组度量可包括定量或分类度量,其代表检测到细胞表面呈递丢失的一种或多种hla蛋白的每一种的一个或多个特征。特别地,转录物组度量可对应于可干扰新抗原呈递的患者特异性肿瘤改变,包括hla突变,hla杂合性丢失和β-2-微球蛋白突变。
89.新抗原呈递预测度量可以通过鉴定利用肿瘤特异性基因组事件(单核苷酸变体,插入或缺失突变以及融合)产生的候选新抗原来产生,所述肿瘤特异性基因组事件利用转录物组数据来验证。所有候选肽可使用用于预测mhc i类呈递的新抗原呈递预测机器学习模型来评分,所述模型可使用大规模免疫肽组数据集来训练。经训练的新抗原呈递预测机器学习模型可使用对应于每一候选肽的数据来产生预测候选肽是否将被呈递和表达于细胞表面上的输出。基于机器学习模型的输出,可以使用通过置信度阈值的候选肽的子集来计算新抗原负荷得分。为了计算综合生物标志物得分,可调整新抗原负荷得分以说明可损害新抗原呈递的受试者特异性肿瘤改变,包括对mhc复合物和抗原呈递机器的改变以及hla的杂合性丢失。c.来源于基因组数据的基因组度量
90.免疫基因组学分析系统可以处理基因组数据以产生一组基因组度量。所述一组基因组度量中的每一个可以代表对应于一条或多条dna序列的相应dna序列的一个或多个特征。在一些情况下,所述一组基因组度量包括:(i)定量或分类度量,其代表一条或多条dna序列中的一个或多个体细胞突变中的每一个的一个或多个特征;(ii)分类度量,其指示在所述生物样品的至少一种人类白细胞抗原(hla)基因中是否已经发生杂合性丢失;和(iii)代表预测的肿瘤突变负荷的定量或分类度量。关于hla的杂合性丢失,可以通过将基因组数据应用于hla缺失-鉴定机器学习模型来产生相应的分类度量。1.单核苷酸变体和插入或缺失突变
91.所述一组基因组度量可包括定量或分类度量,其代表一条或多条dna序列中的一个或多个体细胞突变的每一个的一个或多个特征。所述一个或多个体细胞突变可包括单核苷酸变体,插入/缺失多态性,拷贝数改变和dna序列的一个或多个核酸分子中的融合。在一些情况下,可以为dna序列中每个鉴定的突变产生质量度量,包括突变的数目,转换与颠换
的比率,变体水平一致性等。例如,可以使用质量得分重新校准模块来处理基因组数据,所述质量得分重新校准模块可以通过它们代表假阳性调用的可能性来对单核苷酸变体进行分级。在一些情况下,可以处理基因组数据的序列比对信息,从而可以校正叫错的变体。另外地或可选地,体细胞单核苷酸变体和插入或缺失突变调用可以基于1)比对度量,例如序列覆盖和读取质量,2)位置特征,例如接近空位区域,和3)在正常组织中存在的可能性,通过测试的过滤器集合来组合和分析。2.等位基因特异性hla的杂合性丢失
92.所述一组基因组度量还可以包括分类度量,其指示在生物样品的至少一种hla基因中是否已经发生杂合性丢失。可使用hla缺失-鉴定机器学习模型检测hla的杂合性丢失,因为hla的杂合性丢失可影响新抗原呈递。hla的杂合性丢失可被认为是获得性抗性机制,其通过降低肿瘤新抗原呈递给免疫系统的能力而促进免疫逃逸。由于hla丢失的过程受肿瘤微环境内的选择性压力控制,特别是在肿瘤进化的后期阶段,推测在晚期黑色素瘤受试者群组内,尽管新抗原负荷明显增加,但是等位基因特异性hla的杂合性丢失可有助于降低的治疗响应。
93.为了产生上述基因组度量,可以使用以下步骤处理生物样品:1)将所有肿瘤和正常读数映射至受试者的等位基因特异性hla;2)比对同源等位基因以找到所有患者特异性错配位置;和3)在每个错配位置计算归一化的b-等位基因频率和等位基因特异性覆盖。对于每个基因,将等位基因特异性特征输入到hla缺失-鉴定机器学习模型中以预测杂合性丢失,包括归一化的b等位基因频率和等位基因特异性错配位置,肿瘤纯度和肿瘤倍性。3.突变负荷
94.所述一组基因组度量可包括代表预测的肿瘤突变负荷的定量或分类度量。肿瘤突变负荷可以指在癌细胞的dna中发现的突变(变化)的总数。已知肿瘤突变负荷可有助于计划最佳治疗,并且肿瘤突变负荷已被鉴定为免疫检查点阻断响应的潜在生物标志物。d.产生综合生物标志物得分
95.免疫基因组学分析系统产生从所述一组基因组度量和所述一组转录物度量得到的综合生物标志物得分,并基于综合生物标志物得分确定受试者对特定类型的免疫疗法治疗的响应性的预测水平。例如,综合生物标志物得分可以通过使用对应于新抗原负荷得分的转录物组度量来产生,所述新抗原负荷得分可以基于从基因组数据鉴定的预测的肿瘤突变负荷来调整。因此,综合生物标志物得分可解释对新抗原呈递和其它已建立的抗性标志物的损害。将抗原呈递整合到综合生物标志物得分中可以增强与免疫检查点阻断响应相关的预测水平。
96.尽管提高的新抗原负荷的测量可以预示哪些受试者将受益于免疫疗法,但是综合生物标志物得分可以基于基因组和转录物组度量得到,所述基因组和转录物组度量对应于在种系以及体细胞水平上由抗原呈递机制中的遗传变异引起的另外的抗性机制。这些另外的抗性机制可以通过减少新抗原呈递能力来进一步调节免疫应答。因此,综合生物标志物可以使用对应于新抗原负荷的度量作为生物标志物,但是还可以包括对应于附加数据的基因组和转录物组度量,所述附加数据来源于随后的处理步骤和纵向处理,以及rna表达水平。
97.在一些情况下,综合生物标志物得分对应于新抗原负荷得分,其被调整以解释可
能进一步干扰新抗原呈递的受试者特异性肿瘤改变,包括hla突变,hla的杂合性丢失和β-2-微球蛋白突变。结果,当与新抗原和肿瘤突变负荷分别比较时,使用综合生物标志物得分对受试者的分析可导致改善的对治疗结果的预测。相比在肿瘤免疫应答的更简单的生物模型周围建立的多种当前的生物标志物,模拟生物机制和对新抗原呈递的损害的综合生物标志物方法可以用作免疫检查点阻断疗法的更强的生物标志物。与基于肿瘤突变负荷的方法不同,综合生物标志物得分可以通过模拟新抗原呈递的更广泛机制来产生。
98.另外地或可选地,与对免疫疗法的应答降低相关的体细胞突变的子集(例如,hla i类和b2m突变,hla i类基因中的杂合性丢失)被加权以调整综合生物标志物得分。通过考虑这些逃逸机制,综合生物标志物得分可以捕获呈递给免疫系统的肿瘤抗原的更充分的表示,以增加该生物标志物的预测强度。当应用于一种或多种特定类型的癌症,例如非小细胞肺癌和头颈鳞状细胞癌受试者群组时,上述方法可产生更精确的结果,因为hla的杂合性丢失被鉴定为影响那些类型的癌症进展的普遍逃逸机制。例如,肿瘤数据揭示了在头颈癌,肺腺癌,胰腺癌和前列腺癌中高于45%的频率下的等位基因特异性表达丢失。hla的杂合性丢失,与i类hla基因中体细胞突变的流行相结合,可以通过综合生物标志物得分来捕获,以鉴定对抗原呈递机制的损害事件。
99.因此,综合生物标志物得分可以在多个维度上整合一组广泛的生物特征:外显子组和转录物组,肿瘤和免疫,应答和抗性。综合生物标志物得分然后可用于预测反映驱动对免疫疗法的响应和抗性的生物机制的免疫检查点阻断响应。e.治疗选择
100.综合生物标志物得分可作为免疫检查点阻断疗法响应的强预测因子。如图所示,与肿瘤突变负荷和其它单一分析物/基因,以及发现群组中检查的表达标签相比,综合生物标志物得分实现了免疫检查点阻断疗法响应者和非响应者的更大的分离。用于预测对特定免疫疗法的响应性的综合生物标志物得分的值进一步通过在大的独立验证群组中证实这些发现来证实。
101.综合生物标志物得分可进一步证明,新抗原可指导免疫应答,促进对免疫疗法的临床应答。尽管在应答和肿瘤突变负荷之间仅观察到弱的关联,但是新抗原负荷和受试者应答之间的较强的关联是明显的。已经提出,这种发现可能归因于黑色素瘤亚型在各种临床研究中在患者群组内分布的混淆效应,这对肿瘤突变负荷的预测能力产生负面影响。然而,这种涉及群组的问题似乎不影响新抗原负荷。作为生物标志物的新抗原负荷的增加的稳健性可以通过包括来自后续处理步骤的附加数据以及rna表达水平来实现,因为已经发现这种测量与mhc结合的肽组库中的蛋白质表示相关。
102.在一些情况下,在新抗原负荷外鉴定出影响受试者应答的其它因素。作为说明性实例,在发现群组内,具有观察到的最高综合生物标志物得分的非响应离群值还包括高影响、无义pd-1突变,其可解释为可能阻止对抗pd1疗法的应答。与典型的皮肤黑色素瘤相比,在验证群组中具有高综合生物标志物得分的离群值,非响应受试者对应于患有转移性促结缔组织增生性黑色素瘤的受试者,其与高水平的突变负荷和不同的临床病理学和遗传特征有关。因此,使用临床响应数据以及综合生物标志物得分可以鉴定受试者对免疫疗法的响应的异质性水平。此外,临床响应数据与综合生物标志物得分的组合可以鉴定易受特定疗法组合影响的恶性肿瘤的子集。最后,临床响应数据与综合生物标志物得分的组合可以鉴
定超出新抗原呈递的治疗抗性或响应的其它机制。
103.因此,综合生物标志物得分可用于确定预防,阻止,逆转或改善疾病的治疗方法。所述疾病可以是癌症。综合生物标志物得分可指示受试者的响应性的预测水平。因此,综合生物标志物得分可以作为报告输出,所述报告基于受试者对特定治疗的响应性的预测水平来鉴定:(i)特定治疗的治疗建议;(ii)向人类受试者施用特定治疗的建议;和/或(iii)不向人类受试者施用特定治疗的建议。在一些实施方案中,将土建的治疗施用于人类受试者。
104.癌症的非限制性实例包括:急性成淋巴细胞性白血病、急性髓细胞性白血病、肾上腺皮质癌、aids相关癌症、aids相关淋巴瘤、肛门癌、阑尾癌、室管膜下巨细胞星形细胞瘤(trocytomas)、成神经细胞瘤、基底细胞癌、胆管癌、膀胱癌、骨癌、脑肿瘤如小脑星形细胞瘤、脑星形细胞瘤/恶性神经胶质瘤、室管膜瘤、成神经管细胞瘤、幕上原始神经外胚层肿瘤、视觉通路和下丘脑神经胶质瘤、乳腺癌、支气管腺瘤、伯基特淋巴瘤、未知原始来源的癌、中枢神经系统淋巴瘤、小脑星形细胞瘤、宫颈癌、儿童期癌症、慢性淋巴细胞白血病、慢性骨髓性白血病、慢性骨髓增生性病症、结肠癌、皮肤t细胞淋巴瘤、促结缔组织增生性小圆细胞肿瘤、子宫内膜癌、室管膜瘤、食管癌、尤因肉瘤、生殖细胞肿瘤、胆囊癌、胃癌、胃肠道类癌瘤、胃肠道间质瘤、神经胶质瘤、毛细胞白血病、头颈癌、心脏癌、肝细胞(肝)癌、霍奇金淋巴瘤、下咽癌、眼内黑色素瘤、胰岛细胞癌、卡波西肉瘤、肾癌、喉癌、唇和口腔癌、脂肪肉瘤、肝癌、肺癌、例如非小细胞肺癌和小细胞肺癌、淋巴瘤、白血病、骨/骨肉瘤的巨球蛋白恶性纤维组织细胞瘤、成神经管细胞瘤、黑色素瘤、间皮瘤、具有隐匿性首发口癌的转移性鳞状颈癌、多发性内分泌瘤形成综合征、骨髓增生异常综合征、髓样白血病、鼻腔和副鼻窦癌、鼻咽癌、成神经细胞瘤、非霍奇金淋巴瘤、非小细胞肺癌、口腔癌、口咽癌、骨肉瘤/骨的恶性纤维组织细胞瘤;卵巢癌、卵巢上皮癌、卵巢生殖细胞肿瘤、胰腺癌、胰岛细胞胰腺癌、副鼻窦和鼻腔癌、甲状旁腺癌、阴茎癌、喉癌、嗜铬细胞瘤、松果体星形细胞瘤、松果体生殖细胞瘤、垂体腺瘤、胸膜肺母细胞瘤、浆细胞瘤形成、原发性中枢神经系统淋巴瘤、前列腺癌、直肠癌、肾细胞癌、肾盂和输尿管移行细胞癌、成视网膜细胞瘤、横纹肌肉瘤、唾液腺癌、肉瘤、皮肤癌、梅克尔细胞皮肤癌、小肠癌、软组织肉瘤、鳞状细胞癌、胃癌、t细胞淋巴瘤、喉癌、胸腺瘤、胸腺癌、甲状腺癌、滋养细胞肿瘤(妊娠)、原发部位未知的癌症、尿道癌、子宫肉瘤、阴道癌、外阴癌、waldenstrom巨球蛋白血症和威尔姆氏瘤。可以使用整合的综合生物标志物的疾病或病况的实例包括血液恶性肿瘤、实体瘤恶性肿瘤、转移性癌症和良性肿瘤。
105.患有癌症的各个受试者可以受益于使用整合的综合生物标志物。受试者可以是人、非人灵长类如黑猩猩和其它猿和猴物种;农场动物,例如牛、马、绵羊、山羊、猪;家养动物,例如兔、狗和猫;实验动物包括啮齿动物,例如大鼠、小鼠和豚鼠等。受试者可以是任何年龄的。受试者可以是,例如,老年人、成人、青少年、青春期前儿童、儿童、幼儿、婴儿。
106.患者健康或治疗选择可以通过以下来评估:提供来自受试者的体液或组织样品;从体液或组织样品中收集基因组和蛋白质组谱,并将基因组和蛋白质组谱与至少一种参考谱进行比较以评估受试者的健康。参考谱可以描述以下的至少一种:一种或多种疾病,损伤或病症。参考谱可以由从具有相同疾病的受试者,健康人群或两者收集的基因组或蛋白质组谱建立。所述方法可包括通过随时间反复比较基因组或蛋白质组谱与参考谱来监测。本公开的方面可以包括统计学分析肿瘤谱和参考谱之间的差异以鉴定至少一种生物标志物。可以拒绝显著性水平小于95%、97%、98%或99%的生物标志物或生物标志物组。
wallis h检验。wilcoxon mann-whitney秩和检验(mww)用于数字成对比较。使用benjamini-hochberg校正来调整所列的p值。kolmogorov-smirnov(ks)统计用于rna途径分析。使用kendall's tau确定连续变量之间的相关性。使用逻辑回归产生预测模型,并根据公开的方法使用auroc来确定区分响应和非响应的能力(28)。所有检验均为双侧的;对于途径分析,fdr值《0.1,并且对于所有其它检验,p-值《0.05的被认为是统计学显著的。以下表格提供了b.群组临床数据1.对应于发现群组的样品的临床特征
113.对于在用免疫检查点阻断治疗的群组中的51名不可切除的黑色素瘤受试者,对应于所述群组的中位随访时间为治疗后24个月,其中51名受试者中的33名(50%,95% cloper-pearson置信区间为50-78%)呈现通过实体肿瘤中的响应评估标准(recist)1.1第一次评估的客观响应。在临床群组中,肿瘤起源于头颈区(31%),躯干(31%),手足(25%),肢端区(6%),粘膜(4%)和隐蔽区(2%)。除了这些数据之外,还呈现了性别,年龄和其他受试者特定的人口统计学信息。下表提供了与临床群组的受试者相关的各种特征的概述:表1-发现群组的特征发现群组的特征
114.如表1所示,在疾病来源部位之间的客观响应率方面没有统计学上显著的差异。此外,11名受试者(22%)在先前用检查点抑制剂治疗后进展,而40名(78%)未进行免疫检查点阻断。受试者施用了派姆单抗(n=29,57%)、纳武单抗(n=15,29%)或纳武单抗和伊匹单抗的组合(n=7,14%)。
2.对应于发现群组的样品的基因组数据
115.研究了与响应和非响应肿瘤相关的突变,揭示了在多重假设校正(受试者水平突变数据)后没有明显的响应的单基因预测因子。下表列出了log2和p值,它们为临床群组中提供了响应者和非响应者之间每种鉴定的基因的比较数据:表2
3.对应于群组的受试者的免疫途径数据
116.接着,确定对应于临床数据的遗传破坏的途径。最常被破坏的途径包括rtk-ras和wnt途径(分别在我们的群组的73%和51%中被破坏)。在整个rtk-ras途径中检测到突变。许多rtk被突变,包括ros1和erbb4、ras家族基因,包括nras、braf以及mapk1和2。
117.图2a-b显示了对应于临床群组受试者的基因组和转录物组数据中的致癌变化的统计数据。图2a显示在晚期黑色素瘤受试者中已知致癌途径中的突变。在图2a中,受影响的部分途径表示在途径内突变的基因的数目(n=51个样品,包括在本分析中)。图2b显示在rtk-ras途径内发生的突变的可视化。肿瘤抑制基因以红色列出,并且致癌基因以蓝色显示。点代表特定基因内没有突变。每列代表肿瘤,绿色块代表给定基因内的变体。c.转录物组度量1.差异基因表达
118.为发现群组中的每名受试者产生转录物组数据。从转录物组数据,产生各种转录物组度量。例如,在响应受试者中鉴定出121个差异表达的基因(n=48名可评价的受试者;调整的p值≤0.05,对数倍数变化≥2或≤-0.5)。图3a-c显示了与鉴定与免疫系统应答相关的差异表达基因的转录物组度量相对应的统计数据。图3a显示了群组中具有最高水平差异表达的50个基因,其中提供了倍数变化以比较响应受试者与非响应受试者。图3a进一步显示了48个基因的相应组的每个基因的benjamini-hochberg校正的p值低于0.05。尽管在图3a中没有显示全部,但是在这些基因中的29个当中观察到富集,而在92个基因中观察到表达降低。为了举例说明,图3b显示了临床群组的每名受试者的差异表达基因的热图。在图3b中,每列代表一名受试者,而每行代表一个基因。
119.在最强上调的基因中(log2倍变化=3.28;fdr调整的p=0.0005)包括δ样配体3(dll3),其为在小细胞肺癌和其它肿瘤组织中表现出高表达的抑制性notch配体。由于与在肿瘤中的升高的,均匀的细胞表面表达相比,其在正常组织中的细胞质表达是低的,因此δ-样配体3基因作为可能的治疗靶标目前正在研究中。另外,角蛋白(krt)家族的四个成员(krt72、73、81、86),在比较响应者和非响应者时具有改变的表达水平,所述角蛋白(krt)家族是被鉴定为与癌症发展具有广泛联系的基因组。对dll3和krt家族基因的基因表达分析结果的验证证实了dll3的显著性(mww p=0.02),而krt72、73、81、86没有显著性(mww p=
0.44、p=0.41、p=0.6和p=0.17)。验证结果中的这种差异可能是由于确定单个基因中差异表达的敏感性降低。
120.尽管在群组水平上没有显著富集,但在三名受试者中以非常高的水平检测到ido1表达(中值ido1 tpm=10.36;离群值ido1tpm=1955、661和451)。为了举例说明,图3c显示了比较响应受试者和非响应受试者的ido1基因表达水平的一组框图。基因表达值以每千碱基百万个转录物为单位提供。对于响应受试者组,鉴定了三个离群受试者。尽管ido1表达值似乎与对免疫疗法的响应无关,但这些表达水平升高的离群值可指示在相应受试者中阻止完全响应的逃逸机制(n=48)。例如,过度表达ido1的受试者中的两名可能不能实现对免疫疗法的完全响应,这可能是由于ido1驱动的免疫抑制环境。2.基因富集分析
121.接下来,进行基因集合富集分析以鉴定临床群组中差异调节的途径。图4显示了对应于每个差异调节的免疫途径的归一化富集得分的统计数据,其中归一化富集得分是基于基因集合富集分析产生的。在图4中,在具有上调基因的响应受试者中鉴定了与免疫功能相关的途径的显著富集。显示了0.05以下的benjamini-hochberg校正的p值。炎性信号传导级联是剖析的那些最高度富集的(显著性设置为fdr《0.1)。免疫途径的活化可能是由其它富集的途径引起的。例如,th17的细胞分化可以由以下驱动:(i)细胞因子tgf-β,其在th17细胞中诱导rorγt;和(ii)il-6,其诱导th17谱系。观察到的th17的富集也可以通过观察到的stat3信号传导的增加正调节,所述stat3信号传导用于促进th17分化。3.t细胞受体的表达水平
122.图5a-c显示了与鉴定t细胞受体表达水平的转录物组度量相对应的统计数据。适应性免疫系统由于其独特的t细胞受体(tcr)的大的组库而可响应广泛的抗原。图5a-bc中的框图覆盖了从在其下限的第25个百分位数到在其上限的第75个百分位数的四分位数范围,其中中值由水平线表示。上部线条包括在第75个百分位数以上的1.5x四分位数范围内的最大值。下部线条包括在第25百分位数以下的1.5x四分位数范围内的最小值。为了表征预处理的肿瘤免疫情况,在群组的受试者的子集(n=28个受试者)中鉴定tcr-β组库多样性。使用1-pielou均匀度确定克隆性的所有产生的tcr-β序列的克隆丰度。由于肿瘤内异质性被认为是免疫应答的决定因素,因此将突变型等位基因肿瘤异质性(math)得分与所鉴定的tcr-β克隆性进行比较,所述突变型等位基因肿瘤异质性(math)得分指示肿瘤异质性的估计水平。图5a显示了鉴定低和高突变型等位基因肿瘤异质性水平之间的tcr-β克隆性比较的一组框图。如图5a所示,在高肿瘤异质性和tcrβ组库的克隆多样性之间鉴定出显著的关联(mww,p=0.014)。
123.图5b显示了鉴定第一组受试者和第二组受试者之间tcrβ克隆性比较的一组框图,所述第一组受试者被鉴定为对免疫疗法有响应,所述第二组受试者被鉴定为对免疫疗法无响应。如图5b所示,与非响应者相比,响应受试者的tcr-β克隆性升高(n=28;mww;p=0.047)。因此,tcr-β克隆性可被认为与治疗结果显著相关。此外,图5c显示了鉴定在被鉴定为具有高tcr-β克隆性的第一组和被鉴定为具有低tcr-β克隆性的第二组之间无进展生存概率的比较的线图。图5c显示了与具有低克隆性的受试者相比,在高克隆性受试者中观察到显著更长的无进展生存(双侧km对数秩检验,p=0.0043),其中对于年老/年轻人群(中值群组年龄用作切割点)独立计算高/低分级。
4.免疫浸润标签
124.对群组中肿瘤微环境内的免疫和基质细胞群进行了表征。产生的数据用于产生半定量免疫浸润得分。图6显示了鉴定第一组响应受试者和第二组非响应受试者之间的富集得分的比较的一组框图。通过各种类型的肿瘤浸润淋巴细胞,包括调节性t细胞(treg),天然杀伤细胞(nk细胞)和癌症相关成纤维细胞(caf),来鉴定富集得分的比较。如图6所示,响应和非响应受试者在很大程度上共享不同类型免疫细胞表达的相似分布。因此,单独的免疫细胞的基因表达水平似乎不是对免疫疗法的响应性水平的强预测指标。然而,如本文所述,表达水平可以是产生精确预测对免疫疗法的响应性的综合生物标志物得分的起作用的因素。5.新抗原负荷
125.基于新抗原的生物标志物方法实现了与对免疫检查点阻断的响应的强相关性。对于该特定的示例性实验,产生两种不同的新抗原模型,从而比较它们各自的性能水平。第一新抗原模型对应于仅基于新抗原负荷的得分,而第二新抗原模型对应于第一模型,其被扩展以解释对新抗原呈递和其它已建立的抗性标志物的损伤。因此,第二新抗原模型对应于用于产生综合生物标志物得分的模型。
126.为了计算新抗原负荷得分,使用了来自外显子组和转录物组数据的特征。推定的新表位是从外显子组和转录物组测序两者检测到的单核苷酸变体,插入或缺失突变和融合体预测的。为了改进mhc i类新抗原预测,产生了来自单等位基因hla转染细胞系的基于质谱的肽结合数据。该数据被用于训练一种改进的机器学习算法,所述算法整合了hla结合,蛋白酶体切割和基因表达信息以改进新抗原预测。
127.图7a-b显示了与鉴定跨各种基因和疾病位置的新抗原负荷的转录物组度量相对应的统计数据。图7a显示了鉴定对应于驱动突变的新抗原负荷得分的一组框图,所述驱动突变对应于braf、nras、nf1和wt基因。图7a显示了新抗原负荷在具有不同驱动突变的肿瘤之间显著变化,揭示亚型之间的显著变异(kruskal-wallis,p=1e-04)。
128.此外,图7b显示了鉴定对应于黑色素瘤的各种疾病位置(包括肢端、手足、头/颈、粘膜、躯干和隐蔽区域)的新抗原负荷得分的一组框图。在图7b中,没有检测到疾病来源位置之间的显著关联(kruskal-wallis,p=0.08)。因此,当比较起源于不同来源位置的肿瘤时,新抗原负荷没有全局变化,尽管可以观察到肢端黑色素瘤和躯干黑色素瘤之间的事后比较确实显示出显著的变化(mww;p=0.047)。
129.图8a-f显示了鉴定跨各个受试者的新抗原负荷得分的统计数据,其中新抗原负荷得分可以预测用免疫疗法治疗的受试者的响应性。图8a显示了对应于对免疫疗法响应的第一组受试者和对免疫疗法不响应的第二组受试者之间的新抗原负荷得分的比较的一组框图。在图8a中,每个框图覆盖从其下限的第25个百分位数到其上限的第75个百分位数的四分位数范围,其中中值由水平线表示。上部线条包括在第75个百分位数以上的1.5x四分位数范围内的最大值。下部线条包括在第25百分位数以下的1.5x四分位数范围内的最小值。发现与非响应受试者相比,响应受试者的新抗原负荷显著更高(n=51);mww;p=0.016)。图8b显示了与验证群组(例如,响应受试者,非响应受试者)中受试者组的新抗原负荷得分的比较相对应的一组框图。来自图8b中的验证群组的数据证实,对疗法响应的受试者呈现显著更高的新抗原负荷(mww;p=0.021)。
130.其它类型的实验数据也表明较高的新抗原负荷得分与对免疫疗法的响应性有关。图8c显示了鉴定第一组和第二组之间的无进展生存概率的比较的线图,所述第一组被鉴定为具有高新抗原负荷,所述第二组被鉴定为具有低新抗原负荷。如图8c所示,与具有低新抗原负荷的受试者相比,在具有高新抗原负荷的受试者中观察到显著更长的无进展生存(双侧km对数秩检验;p=0.002)。图8d显示了鉴定验证群组中的受试者组之间的无进展生存概率的比较的线图,而图8e显示了鉴定验证群组中的受试者组之间的总体生存率的比较的线图。尽管图8d显示在验证群组中具有高新抗原负荷的受试者的无进展生存没有显著长于具有低新抗原负荷的受试者(双侧km对数秩检验,p=0.085),但是图8e显示了在具有高新抗原负荷的受试者中观察到总体生存的显著改善(双侧km对数秩检验,p=0.085)。
131.图8f显示了鉴定新抗原负荷得分模型的性能水平的接受者操作特征曲线。如图8f所示,新抗原负荷得分模型的曲线下面积值为0.71,交叉验证曲线下面积值(平均值)为0.69(对数似然比p=0.0329)。d.基因组度量1.突变特征
132.除了转录物组数据之外,还为发现群组中的每名受试者产生基因组数据。从基因组数据产生各种基因组度量。图9a-f显示了鉴定与发现群组的每个受试者样品中存在的突变相关的一个或多个特征的统计数据。图9a显示了接受抗pd-1疗法的受试者的各种基因中的突变。在图9a中,框图表示突变负载。平铺图显示了逐个样品(列)的突变基因(行),其中平铺颜色指示突变类型。右边的框图代表在特定基因中具有突变的受试者的数目,其被着色以指示突变类型。在平铺图下,第一条线代表治疗响应,作为响应(部分或完全响应;深绿色;n=33),或非响应(黑色;n=18)。
133.在图9a中,中值非同义肿瘤突变负荷为4.07个突变/mb(四分位数范围,0.95-12.455)。该基因组度量似乎与已知数据集中观察到的值一致。例如,图9b显示了跨各种数据集在每个样品中鉴定的突变的量。发现群组中的突变负荷水平与tcga-skcm数据集(黑色素瘤)中的那些相当。在图9b中,每个点代表样品,在每种癌症类型中,红色水平线处于突变的中值数。垂直轴(对数尺度)表示每个样品的突变数目。
134.图9c显示了鉴定每种类型的单核苷酸变体的突变量的一组框图和显示在发现群组中每名受试者的单核苷酸变体类型的分布的条形图。在图9c中,单核苷酸变体被分类为转换或颠换(n=49)。左侧框图显示了六种不同取代类型的总体分布,而右侧框图显示了转换(t1)和颠换(tv)的分布。如图9c所示,c》t转换似乎形成鉴定的单核苷酸变体的大部分(76%)。
135.图9d显示了鉴定发现群组中每名受试者的三个突变标签的分布的条形图。通过分解核苷酸取代的矩阵来提取标签,基于紧围绕突变碱基的碱基将其分类为96个取代类别,从而在群组内产生三个主要标签。根据图9d,在33%的受试者中,在braf中发生最普遍鉴定的驱动突变,随后在人群中发生20%的nras和16%的nf1。图9e显示了三个主要标签的突变的分布。将所提取的标签与先前验证的标签进行比较。发现群组中的标签1和2与uv标签最相似,而第三标签与未知病因的标签最密切相关。如图9e所示,发现群组中发现的突变标签与uv诱导的dna损伤最密切相关。
136.图9f显示了针对与特定肿瘤相关的每种驱动突变(例如,braf、nras)鉴定对应于
对免疫疗法的各种水平的响应性的受试者分布的条形图。例如,响应者被定义为完全响应(cr)或部分响应(pr)。非响应者被定义为稳定疾病(sd)或进行性疾病(pd)。驱动突变可指为癌细胞的肿瘤转化赋予基本生长优势的基因改变。在图9f中,具有braf突变肿瘤的受试者更可能对疗法有积极响应(n=47;精确二项式检验;p=0.0258)。不同基因组亚型的响应率与预期的响应率没有显著差异。受试者中wt基因的进行性疾病的数目增加可能是由braf的频率降低(其通常以较高的频率被观察到)引起的。2.肿瘤突变负荷
137.图10显示了鉴定跨各种驱动突变、疾病位置和受试者组的肿瘤突变负荷的多组框图1000。框图1000包括框图1002、1004和1006。框图覆盖从其下限的第25个百分位数到其上限的第75个百分位数的四分位数范围,其中中值由水平线表示。上部线条包括在第75个百分位数以上的1.5x四分位数范围内的最大值。下部线条包括在第25百分位数以下的1.5x四分位数范围内的最小值。对应于肿瘤突变负荷的值以log10尺度绘制。
138.框图1002鉴定每种驱动突变的肿瘤突变负荷。肿瘤突变负荷在具有不同驱动突变的肿瘤之间显著变化(kruskal-wallis,p=0.00012)。框图1004鉴定黑色素瘤的疾病来源的每个鉴定位置的肿瘤突变负荷。框图1004显示跨疾病来源的不同部位的肿瘤突变负荷的显著全局变化,其中与头和颈来源的黑色素瘤相比发现显著变化(kruskal-wallis,p=0.016)。
139.框图1006鉴定对免疫疗法响应的第一组受试者和对免疫疗法没有响应的第二组受试者的肿瘤突变负荷。肿瘤突变负荷在响应和非响应受试者中的比较显示显著的关联(mmw;p=0.049)。然而,在该群组中响应和非响应受试者的肿瘤突变负荷之间相对较小的差异可能是由于黑色素瘤亚型的混淆效应和变化的肿瘤纯度,因为最近已经表明这些测量限制了肿瘤突变负荷作为预测生物标志物的有效性。因此,单独的肿瘤突变负荷可能不能精确地预测对免疫疗法的响应性。e.综合生物标志物得分
140.如本文所述,本公开的实施方案认识到抗原呈递机制中的改变可干扰新抗原呈递。考虑这些数据可以改善预测对免疫疗法的响应性的性能,因为这些改变已经单独被注意到影响受试者对免疫检查点阻断的响应。因此,综合生物标志物得分调整新抗原负荷得分以解释能够干扰新抗原呈递的受试者特异性肿瘤改变,包括hla突变,hla的杂合性丢失和b2m突变。1.发现群组
141.图11a-d显示了鉴定跨各个受试者的综合生物标志物得分的统计数据,其中综合生物标志物得分表明在预测用免疫疗法治疗的受试者的响应性方面的表现改善。具体地,图11a-d显示了综合生物标志物得分比单独的新抗原负荷与对免疫疗法的响应更强地相关。例如,图11a显示了与对免疫疗法有响应的第一组受试者和对免疫疗法没有响应的第二组受试者之间的综合生物标志物得分的比较相对应的一组框图。如图11a所示,与非响应受试者相比,响应受试者中的综合生物标志物得分显著更高(n=51;mww;p=0.002)。因此,当与新抗原负荷相比时,综合生物标志物得分导致改善的对治疗结果的预测。图11b显示了与验证群组中受试者组(例如,响应受试者,非响应受试者)的综合生物标志物得分的比较相对应的一组框图。来自图11b中的验证群组的数据证实了类似的结果,其中响应组中的受试
者呈现比非响应受试者显著更高的综合生物标志物得分(n=110;mww;p=0.010)。参考图11a-b,相应的框图覆盖从其下限的第25个百分位数到其上限的第75个百分位数的四分位数范围,其中中值由水平线表示。上部线条包括在第75个百分位数以上的1.5x四分位数范围内的最大值。下部线条包括在第25百分位数以下的1.5x四分位数范围内的最小值。
142.图11c显示了鉴定第一组和第二组之间的无进展生存概率的比较的线图,所述第一组被鉴定为具有高综合生物标志物得分,所述第二组被鉴定为具有低综合生物标志物得分。如图11c所示,与具有低综合生物标志物得分的受试者相比,在具有高综合生物标志物得分的受试者中观察到显著更长的无进展生存(双侧km对数秩检验;p=0.0016)。
143.图11d显示了鉴定综合生物标志物得分模型的性能水平的接受者操作特征曲线。如图11d所示,综合生物标志物模型比新抗原负荷模型表现得更好:综合生物标志物得分的曲线下面积从0.71增加到0.76,并且交叉验证曲线下面积(平均值)从0.69增加到0.75(对数似然比p=0.0057)。2.验证群组
144.图12a-b显示了鉴定跨各个受试者的综合生物标志物得分的统计数据,其中综合生物标志物得分表明在预测群组中受试者的无进展和总体生存率方面的表现改善。具体地,综合生物标志物得分的性能水平的改善在验证群组中更显著。图12a显示了鉴定验证群组中的受试者组之间的无进展生存概率的比较的线图,而图12b显示了鉴定验证群组中的受试者组之间的总体生存率的比较的线图。与在验证群组中发现的新抗原负荷得分相反,图12a显示了与高综合生物标志物得分相关的受试者的无进展生存显著长于与低综合生物标志物得分相关的受试者(双侧km对数秩检验,p=0.05)。还如图12b所示,当分析总体生存时,也获得了更大的显著性,其中在与高综合生物标志物得分相关的受试者中的总体生存率显著更长(双侧km对数秩检验,p=0.002)。综合生物标志物得分的改善可在生物学上理解为发现群组中23.5%的受试者和验证群组中17.27%的受试者具有至少一种潜在地影响抗原呈递的机制这样的发现,这提示这些特征可经常影响免疫系统对免疫疗法的应答。3.影响综合生物标志物得分的hla基因突变
145.图13a-b显示了鉴定hla基因的体细胞突变的统计数据,所述hla基因的体细胞突变可能有助于降低新抗原呈递的概率。具体地,对跨发现群组的hla突变的损害的综述揭示了许多受试者中的有害变体。例如,图13a显示了在发现群组的样品中鉴定的体细胞变体的实例。如图13a所示,在受试者25中发现了两种不同的体细胞hla突变,包括hla-a02:01中的停止获得突变和hla-b15:01中的剪接区变体(等位基因分数分别=0.473和0.368)。这些体细胞突变可导致hla-a02:01表面表达的丢失和hlab15:01可能的错误折叠。在受试者38的β-2-微球蛋白(b2m)中检测到破坏性移码变体,其可能损害该受试者中所有的mhc i类呈递。
146.图13b显示了鉴定新抗原的相对频率的条形图,所述新抗原由发现群组的受试者25的相应hla基因呈现。在图13b中,预测受试者25中38.9%的新抗原(对于a02:01为19.1%;对于b15:01为19.8%)与受损的hla等位基因结合,提示对新抗原呈递的潜在严重损害。值得注意的是,受试者25在非响应受试者中是离群值,具有高得多的新抗原负荷,提示除了在综合生物标志物得分中捕获的新抗原外,受损的新抗原呈递也可能是免疫检查点阻断抗性的起作用的因素。在另一个离散受试者38(高新抗原负荷,无响应者)中,以高等位
基因分数检测到b2m中的破坏性移码变体,其也潜在地影响抗原呈递。4.hla的杂合性丢失
147.在该群组中还检测到hla的杂合性丢失,因为它也可影响新抗原的呈递。hla的杂合性丢失是指通过降低肿瘤新抗原呈递给免疫系统的能力而促进免疫逃逸的获得性抗性机制。由于hla丢失的过程受肿瘤微环境内的选择性压力控制,特别是在肿瘤发展的后期,假设在晚期黑色素瘤受试者群组内,等位基因特异性hla的杂合性丢失可有助于降低治疗响应,尽管新抗原负荷明显增加。
148.发现hla的杂合性丢失是hla破坏的最常见形式,发生在19.6%的可评价受试者(10/51)中,其中三个个体在所有非纯合hla中呈现杂合性丢失。图14a-b显示了鉴定特定受试者的正常样品和相应肿瘤样品之间的hla序列的比较的一组图的实例。例如,图14a显示了鉴定受试者的正常和肿瘤样品之间的hla-a序列的比较的一组图,而图14b显示了鉴定受试者的正常和肿瘤样品之间的hla-c序列的比较的一组图。
149.图14a-b的图提供了发现群组的受试者54的hla-a和hla-c中hla的杂合性丢失的基于ngs序列的证据。未显示hla-b。第一行显示了正常样品中两个同源等位基因的原始读数覆盖。第二行显示了肿瘤样品中两个同源等位基因的原始读数覆盖。两幅图都具有代表两个等位基因之间的差异位置的垂直灰线。由于严格的映射参数要求所有的读数无错配地映射,因此灰色线处的覆盖差异代表等位基因之间的真实覆盖差异。第三幅图显示了来自正常样品(灰色)和肿瘤样品(黑色)的b-等位基因频率。肿瘤样品中的b-等位基因频率应根据正常样品中的b-等位基因频率考虑,因为等位基因之间的引物杂交差异。第四幅图显示了肿瘤和正常样品之间每个等位基因的覆盖率。这些值已经通过肿瘤和正常读数深度跨整个外显子组而归一化。没有拷贝数变化的预期值是1,用灰色虚线显示。第三幅和第四幅图仅显示两个等位基因之间错配位置的数据。
150.如图14a-b所示,来自受试者的匹配的正常组织通常表现出跨越hla基因a和c的均匀等位基因特异性覆盖。相反,来自该受试者的肿瘤组织在跨越每个hla的大部分的等位基因特异性覆盖中表现出广泛的不平衡,其中在hla-a01:01和hla-c07:01中的覆盖水平低。b-等位基因频率(b-等位基因频率)显示与正常的绝对差异。在丢失的等位基因中观察到始终较低的覆盖率(图14a-b中的第四行),预测其呈递约54%的该受试者的新抗原,可能降低呈现给免疫系统的能力。vii.用于产生综合生物标志物得分的方法
151.图15包括说明根据一些实施方案产生综合生物标志物得分的方法的实例的流程图1500。流程图1500中描述的操作可以由例如实施一个或多个操作的计算机系统来执行,所述操作用于基于转录物组和基因组度量生成综合生物标志物得分。尽管流程图1500可以将操作描述为顺序过程,但是在各种实施方案中,可以并行或同时地执行许多操作。此外,可以重新安排操作的顺序。操作可以具有图中未显示的其他步骤。此外,所述方法的实施方案可以通过硬件,软件,固件,中间件,微代码,硬件描述语言或其任何组合来实施。当在软件,固件,中间件或微代码中实施时,用于执行相关任务的程序代码或代码段可以存储在诸如存储介质的计算机可读介质中。
152.在操作1510,免疫基因组学分析系统访问通过处理受试者的生物样品生成的基因组数据和转录物组数据。在一些情况下,生物样品包括一种或多种癌细胞。基因组数据可鉴
定生物样品中的一条或多条dna序列,其中可进行全外显子组测序以鉴定一条或多条dna序列。转录物组数据可鉴定生物样品中的一条或多条rna序列,其中转录物组测序可用于鉴定一条或多条rna序列。另外地或可选地,基因组和转录物组数据可以从包括受试者的生物样品和参考生物样品的样品对产生,其中参考生物样品不包括一种或多种癌细胞。
153.在操作1520,免疫基因组学分析系统处理基因组数据以生成一组基因组度量。所述一组基因组度量中的每一个可以代表对应于一条或多条dna序列的相应dna序列的一个或多个特征。在一些情况下,所述一组基因组度量包括:(i)定量或分类度量,其代表一条或多条dna序列中的一个或多个体细胞突变中的每一个的一个或多个特征;(ii)分类度量,其指示在所述生物样品的至少一种人类白细胞抗原(hla)基因中是否已经发生杂合性丢失;和(iii)代表预测的肿瘤突变负荷的定量或分类度量。关于hla的杂合性丢失,可以通过将基因组数据应用于hla缺失-鉴定机器学习模型来产生相应的分类度量。
154.在操作1530,免疫基因组学分析系统处理转录物组数据以生成一组转录物组度量。所述一组转录物组度量中的每一个可代表对应于一组肽的一个或多个特征,所述一组肽由一条或多条rna序列的相应rna序列翻译而成。在一些情况下,所述一组转录物组度量包括:(i)定量或分类度量,其代表生物样品的预测的新抗原负荷;(ii)定量或分类度量,其代表从所述生物样品检测到的一种或多种候选新抗原中的每一种的一个或多个特征;(iii)定量或分类度量,其代表检测到细胞表面呈递丢失的一种或多种hla蛋白中的每一种的一个或多个特征;(iv)定量或分类度量,其代表对应于hla基因的一个或多个特征,所述hla基因编码检测到细胞表面呈递丢失的一种或多种hla蛋白;(v)定量或分类度量,其代表对应于免疫细胞的序列的表达水平;和(vi)定量或分类度量,其代表从所述生物样品检测到的一种或多种t细胞受体的表达水平。对于检测到细胞表面呈递丢失的hla蛋白,可以通过将基因组和转录物组数据应用于新抗原呈递预测机器学习模型来产生相应的度量。
155.在操作1540,免疫基因组学分析系统生成来源于所述的一组基因组度量和所述的一组转录物组度量的综合生物标志物得分。在一些情况下,免疫基因组学分析系统通过以下步骤生成综合生物标志物得分:(i)将一组基因组度量中的每个基因组度量与基于一组转录物组度量中相应的转录物组度量确定的权重值进行加权;和(ii)使用加权的基因组度量生成综合生物标志物得分。
156.在操作1550,免疫基因组学分析系统基于综合生物标志物得分,确定受试者对特定类型的免疫疗法治疗的响应性的预测水平。
157.在操作1560,免疫基因组学分析系统输出对应于受试者的响应性的预测水平的结果。结果可以是基于受试者对特定治疗的响应性的预测水平来鉴定以下的报告:(i)特定治疗的治疗建议;(ii)向人类受试者施用特定治疗的建议;和/或(iii)不向人类受试者施用特定治疗的建议。在一些实施方案中,将推荐的治疗施用于人类受试者。此后,过程1500终止。viii.其他考虑因素
158.虽然本发明的主题已经结合其具体实施方案进行了详细描述,但是应当理解,本领域技术人员在理解上述内容后,可以容易地对这些实施方案进行改变,变化和产生其等同物。因此,应当理解,本公开是出于示例而非限制的目的而呈现的,并且不排除包括如本领域技术人员将容易明白的对本主题的这些修改,变化和/或添加。实际上,本文描述的方
法和系统可以以各种其它形式来体现。此外,在不脱离本公开的精神的情况下,可以在本文描述的方法和系统的形式上进行各种省略,替换和改变。所附权利要求及其等同物旨在覆盖落入本公开的范围和精神内的这些形式或修改。
159.除非另有具体说明,否则应当理解,在本说明书中,使用诸如“处理”、“计算(computing)”、“计算(calculating)”、“确定”和“鉴定”等术语的讨论是指计算装置(诸如一个或多个计算机或类似的一个或多个电子计算装置)中操纵或转换呈现为计算平台的存储器、暂存器、或其它信息存储装置、传输装置或显示装置内的物理电子或磁量的数据的动作或过程。
160.本文讨论的一个或多个系统不限于任何特定的硬件结构或配置。计算装置可以包括提供基于一个或多个输入调节的结果的组件的任何合适的布置。合适的计算装置包括访问存储的软件的多用途的基于微处理器的计算系统,所述存储的软件将计算系统从通用目的计算设备编程或配置到实施本发明的主题的一个或多个实施方案的专用计算设备。可以使用任何合适的编程,脚本或其它类型的语言或语言的组合来在编程或配置计算装置中使用的软件中实施本文所包含的教导。
161.一些实施方案的实施方案可以在这种计算装置的操作中执行。在上述示例中呈现的模块的顺序可以改变,例如,模块可以被重新排序,组合和/或分割成子块。某些模块或过程可以并行执行。
162.除非在所使用的上下文中另外具体说明或另外理解,否则本文所使用的条件语言(例如“可以”、“能够”、“可能”、“可以”、“例如”等)通常旨在传达某些实例包括某些特征、元件和/或步骤,而其他实例不包括某些特征、元件和/或步骤。因此,这种条件语言通常并非旨在暗示特征,元件和/或步骤以任何方式为一个或多个实例所必需的,或者一个或多个实例必然包括用于在有或没有作者输入或提示的情况下决定这些特征,元件和/或步骤是否被包括在或要在任何特定实例中执行的逻辑。
163.术语“包括”、“包含”、“具有”等是同义的,并且以开放式的方式被包含地使用,并且不排除附加的元件、特征、动作、操作等。此外,术语“或”以其包含的意义(而不是以其排他的意义)使用,使得当例如用于连接元件列表时,术语“或”意味着列表中的一个、一些或所有元件。本文使用的“适于”或“被配置成”意味着开放和包括在内的语言,其不排除适于或被配置成执行另外的任务或步骤的装置。另外,“基于”的使用意味着是开放的和包括在内的,因为“基于”一个或多个所述条件或值的过程,步骤,计算或其它动作在实践中可以基于超出那些所述条件或值的附加条件或值。类似地,“至少部分地基于”的使用意味着是开放的和包括在内的,因为“至少部分地基于”一个或多个所述条件或值的过程,步骤,计算或其它动作在实践中可以基于超出那些所述条件或值的附加条件或值。本文包括的标题,列表和编号仅仅是为了便于解释,并不意味着是限制性的。
164.上述各种特征和过程可以彼此独立地使用,或者可以以各种方式组合使用。所有可能的组合和子组合旨在落入本公开的范围内。此外,在一些实施方式中可以省略某些方法或过程模块。本文描述的方法和过程也不限于任何特定顺序,并且可以以适当的其它顺序来执行与其相关的模块或状态。例如,所描述的模块或状态可以以不同于具体公开的顺序来执行,或者可以将多个模块或状态组合在单个模块或状态中。示例模块或状态可以串行,并行或以某种其它方式执行。可以将模块或状态添加到所公开的实例中或从所公开的
实例中移除。类似地,本文描述的示例系统和组件可以以不同于所述的方式来配置。例如,与所公开的实例相比,可以将元件添加到所公开的实例中,从所公开的实例中移除或重新排列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1