树脂组合物的制作方法
1.本发明涉及含有聚酰亚胺前体/聚酰亚胺的树脂组合物和聚酰亚胺薄膜。本发明还涉及含有聚酰亚胺前体/聚酰亚胺的树脂组合物的制造方法、以及聚酰亚胺薄膜、显示器、层叠体和柔性设备的制造方法。
背景技术:
2.聚酰亚胺树脂是不溶解、不熔融的超耐热性树脂,具有耐热氧化性、耐热特性、耐辐射线性、耐低温性、耐化学药品性等优异的特性。因此,聚酰亚胺树脂在包括电子材料在内的广泛领域中使用。作为聚酰亚胺树脂在电子材料领域中的应用例,可列举出例如绝缘涂布材料、绝缘膜、半导体、薄膜晶体管液晶显示器(tft-lcd)的电极保护膜等。最近,利用聚酰亚胺薄膜的轻便、柔软性等,研究了将其替代以往在显示器材料的领域中使用的玻璃基板而用作柔性基板。
3.专利文献1中记载了一种聚酰亚胺薄膜,其如下获得:使用3,5-二氨基苯甲酰胺(以下也称为daba)、下述结构式的化合物(以下也称为cpoda)和其它化合物作为单体来合成聚酰亚胺,向所得聚酰亚胺清漆中添加交联剂,进行涂布、干燥而得到。
[0004][0005]
另外,专利文献1中还记载了该薄膜的气体分离性能优异。专利文献2中记载了一种透明聚酰亚胺薄膜,其是将使用daba、tfmb(2,2
’‑
双(三氟甲基)联苯胺)等作为二胺且使用cpoda作为酸二酐的聚酰亚胺清漆进行涂布、干燥而得到的。
[0006]
现有技术文献
[0007]
专利文献
[0008]
专利文献1:日本特许第6551640号公报
[0009]
专利文献2:国际公开第2019/211972号
技术实现要素:
[0010]
发明要解决的问题
[0011]
专利文献1中记载的聚酰亚胺薄膜的用途为气体分离膜,并未作为光学材料而进行研究。
[0012]
另外,本发明人等发现:将daba、tfmb和cpoda、含硅化合物等用作单体,与上述专利文献2中记载的内容同样地合成聚酰亚胺清漆时,在清漆制造工艺中寻求的清漆过滤性不充分,将该聚酰亚胺清漆进行涂布、加热而得到的聚酰亚胺薄膜的在显示器用途中寻求的特性(拉伸伸长率等)不充分。另一方面,本发明人等发现:将daba、3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)、9,9-双(4-氨基苯基)芴(bafl)等二胺、cpoda、4,4
’‑
氧双邻苯二甲酸酐(odpa)、9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)等酸二酐和其它
含硅化合物等用作单体而得到的聚酰亚胺的在显示器用途中寻求的特性优异。
[0013]
因此,本发明是鉴于上述情况而进行的,其目的在于,提供在含有聚酰亚胺前体和/或聚酰亚胺的树脂组合物的制造工艺中寻求的特性以及在显示器用途中寻求的其它特性这两者优异的聚酰亚胺薄膜,以及用于形成其的树脂组合物。
[0014]
用于解决问题的方案
[0015]
上述课题可通过以下的技术手段来解决。
[0016]
〔第一方式〕
[0017]
《1》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0018][0019]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0020][0021]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0022]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0023][0024]
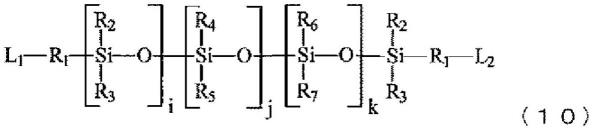
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0025]
并且,所述树脂组合物以前述树脂的总质量作为基准包含25质量%以下的前述含硅化合物。
[0026]
《2》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0027][0028]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0029][0030]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0031]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0032][0033]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0034]
并且,前述树脂的酰亚胺化率为50%以上。
[0035]
《3》根据项目1所述的树脂组合物,其中,前述树脂的酰亚胺化率为50%以上。
[0036]
《4》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0037][0038]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0039][0040]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0041]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0042][0043]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各
自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0044]
并且,所述树脂组合物满足下述(一)或(二)中的任意者,
[0045]
(一)前述p3和/或p5包含源自下述通式(8)所示化合物的结构单元,
[0046]
或者
[0047]
(二)前述p4和/或p6包含源自下述通式(9)所示化合物的结构单元,
[0048][0049]
《5》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0050][0051]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0052][0053]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0054]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0055][0056]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0057]
并且,前述p3和/或p5各自独立地包含至少一个源自3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)或9,9-双(4-氨基苯基)芴(bafl)中的各种化合物
的结构单元。
[0058]
《6》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0059][0060]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0061][0062]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0063]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0064][0065]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0066]
并且,前述p3和/或p5各自独立地包含至少一个源自2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)的结构单元。
[0067]
《7》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0068][0069]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0070][0071]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0072]
前述p5或p6包含源自下述通式(10)所示含硅化合物的结构单元,
[0073][0074]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0075]
并且,将a(质量%)设为构成通式(7)中的p5的二胺之中的通式(10)的比例,将b(质量%)设为构成通式(6)中的p3的二胺之中的通式(10)的比例时,b-a大于0且小于60。
[0076]
《8》根据项目7所述的树脂组合物,其中,前述二胺为选自下述通式(8)所示的化合物、3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)和9,9-双(4-氨基苯基)芴(bafl)中的至少一种化合物。
[0077][0078]
《9》根据项目1~8中任一项所述的树脂组合物,其中,前述通式(6)和/或前述通式(7)中,p4和/或p6各自独立地包含至少一个源自4,4
’‑
氧双邻苯二甲酸酐(odpa)或9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)中的各种化合物的结构单元。
[0079]
《10》根据项目1~9中任一项所述的树脂组合物,其中,前述通式(10)所示的含硅化合物的官能团当量为800以上。
[0080]
《11》根据项目1~10中任一项所述的树脂组合物,其中,
[0081]
前述p5包含源自前述通式(10)所示化合物的结构单元,且
[0082]
前述通式(10)中,前述l1和l2各自独立地为氨基。
[0083]
《12》根据项目4和9~11中任一项所述的树脂组合物,其中,将全部二胺(不包括前述通式(10)所示的化合物)设为100mol%时,前述通式(8)所示的化合物多于50mol%。
[0084]
《13》根据项目1~12中任一项所述的树脂组合物,其中,前述p3或p5各自独立地包含源自下述式所示化合物(bisam)的结构单元。
[0085][0086]
《14》根据项目1~13中任一项所述的树脂组合物,其中,前述p4或p6各自独立地包含源自4,4
’‑
(六氟异亚丙基)双邻苯二甲酸二酐、下述式所示的化合物(bzda)、
[0087][0088]
或者下述式所示的化合物(bnbda)
[0089][0090]
的结构单元。
[0091]
《15》根据项目1~14中任一项所述的树脂组合物,其中,将前述树脂加热而得到的聚酰亚胺树脂膜被用于柔性基板。
[0092]
《16》根据项目1~15中任一项所述的树脂组合物,其中,将前述树脂固化而得到的聚酰亚胺树脂膜被用于柔性显示器。
[0093]
《17》一种树脂组合物的制造方法,其包括:
[0094]
使二胺或酸二酐与下述通式(10)所示的含硅化合物发生缩聚反应而得到聚酰亚胺后,
[0095]
使其与其它化合物发生缩聚反应,提供包含树脂的树脂组合物,所述树脂包含聚酰亚胺前体和聚酰亚胺,
[0096][0097]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0098]
所述树脂组合物以前述树脂的总质量作为基准包含25质量%以下的前述含硅化合物。
[0099]
《18》一种树脂组合物的制造方法,其包括:
[0100]
使二胺或酸二酐与下述通式(10)所示的含硅化合物发生缩聚反应而得到聚酰亚胺后,
[0101]
使其与其它化合物发生缩聚反应,提供包含树脂的树脂组合物,所述树脂包含聚酰亚胺前体和聚酰亚胺,
[0102][0103]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0104]
前述树脂的酰亚胺化率为50%以上。
[0105]
《19》一种树脂组合物的制造方法,其包括:
[0106]
使下述通式(8)所示的化合物或下述通式(9)所示的化合物与下述通式(10)所示的含硅化合物与其它化合物发生缩聚反应而得到聚酰亚胺后,
[0107]
使其与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物。
[0108][0109]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0110]
《20》一种树脂组合物的制造方法,其包括:
[0111]
使选自3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)和9,9-双(4-氨基苯基)芴(bafl)中的至少一种化合物与下述通式(10)所示的含硅化合物与其它化合物发生缩聚反应而得到聚酰亚胺后,
[0112]
使其与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物。
[0113][0114]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0115]
《21》一种树脂组合物的制造方法,其包括:
[0116]
使选自4,4
’‑
氧双邻苯二甲酸酐(odpa)和9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)中的至少一种化合物与下述通式(10)所示的含硅化合物与其它化合物发生缩聚反应而得到聚酰亚胺后,
[0117]
使其与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物。
[0118][0119]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0120]
《22》一种聚酰亚胺树脂膜的制造方法,其包括:
[0121]
在支承体的表面上涂布项目1~16中任一项所述的树脂组合物或通过项目17~21中任一项所述的方法而得到的树脂组合物的涂布工序;
[0122]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及
[0123]
将该聚酰亚胺树脂膜从该支承体上剥离的剥离工序。
[0124]
《23》根据项目22所述的聚酰亚胺树脂膜的制造方法,其包括:
[0125]
在前述剥离工序之前,从前述支承体侧对前述树脂组合物照射激光的照射工序。
[0126]
《24》一种显示器的制造方法,其包括:
[0127]
在支承体的表面上涂布项目1~16中任一项所述的树脂组合物或通过项目17~21中任一项所述的方法而得到的树脂组合物的涂布工序;
[0128]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;
[0129]
在该聚酰亚胺树脂膜上形成元件的元件形成工序;以及
[0130]
将形成有该元件的该聚酰亚胺树脂膜从该支承体上剥离的剥离工序。
[0131]
《25》一种层叠体的制造方法,其包括:
[0132]
在支承体的表面上涂布项目1~16中任一项所述的树脂组合物或通过项目17~21中任一项所述的方法而得到的树脂组合物的涂布工序;
[0133]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及
[0134]
在该聚酰亚胺树脂膜上形成元件的元件形成工序。
[0135]
《26》根据项目25所述的层叠体的制造方法,其还包括:
[0136]
将形成有前述元件的前述聚酰亚胺树脂膜从前述支承体上剥离的工序。
[0137]
《27》一种柔性设备的制造方法,其包括:
[0138]
通过项目25或26所述的方法来制造层叠体。
[0139]
〔第二方式〕
[0140]
《28》一种树脂组合物,其包含下述通式(1)或(2)所示结构单元的树脂,
[0141][0142]
{式中,p1表示二价有机基团;p2表示四价有机基团;并且,p表示正整数。}
[0143][0144]
{式中,p1表示二价有机基团;p2表示四价有机基团,并且,p表示正整数。}
[0145]
前述p1包含源自下述通式(3)所示化合物的结构单元,
[0146][0147]
并且,前述p1或p2包含源自下述通式(5)所示含硅化合物的结构单元,
[0148][0149]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50;且官能团当量为800以上。}
[0150]
《29》根据项目28所述的树脂组合物,其中,前述p2包含源自下述通式(4)所示化合物的结构单元。
[0151][0152]
《30》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0153][0154]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0155][0156]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0157]
前述p3或p4包含源自下述通式(10)所示含硅化合物的结构单元。
[0158][0159]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0160]
《31》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0161][0162]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0163][0164]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0165]
前述p3或p4包含源自下述通式(10)所示含硅化合物的结构单元,
[0166][0167]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0168]
并且,所述树脂组合物满足下述(一)或(二)中的任意者,
[0169]
(一)前述p3和/或p5包含源自下述通式(8)所示化合物的结构单元,
[0170]
或者
[0171]
(二)前述p4和/或p6包含源自下述通式(9)所示化合物的结构单元。
[0172][0173]
《32》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0174][0175]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0176][0177]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0178]
前述p3或p4包含源自下述通式(10)所示含硅化合物的结构单元,
[0179][0180]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0181]
并且,前述p3和/或p5包含至少一个源自3,3
’‑
二氨基二苯基砜(33das)或4,4
’‑
二氨基二苯基砜(44das)中的各种化合物的结构单元。
[0182]
《33》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0183][0184]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0185][0186]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0187]
前述p3或p4包含源自下述通式(10)所示含硅化合物的结构单元,
[0188][0189]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0190]
并且,前述p3和/或p5包含至少一个源自9,9-双(4-氨基苯基)芴(bafl)或下述通式的化合物(bisam)中的各种化合物的结构单元。
[0191][0192]
《34》一种树脂组合物,其包含下述通式(6)和(7)所示结构单元的树脂,
[0193][0194]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0195][0196]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)。}
[0197]
前述p3或p4包含源自下述通式(10)所示含硅化合物的结构单元,
[0198][0199]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0200]
并且,前述p4和/或p6包含至少一个源自
[0201]
4,4
’‑
氧双邻苯二甲酸酐(odpa)、
[0202]
4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐(6fda)、
[0203]
9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)、
[0204]
下述通式的化合物(bzda)、
[0205][0206]
或者下述通式的化合物(bnbda)
[0207][0208]
中的各种化合物的结构单元。
[0209]
《35》根据项目30~34中任一项所述的树脂组合物,其中,
[0210]
前述p3包含源自前述通式(10)所示化合物的结构单元,
[0211]
前述通式(10)所示的含硅化合物的官能团当量为800以上。
[0212]
《36》根据项目30~35中任一项所述的树脂组合物,其中,
[0213]
前述p3包含源自前述通式(10)所示化合物的结构单元,且
[0214]
前述通式(10)中,前述l1和l2各自独立地为氨基。
[0215]
《37》根据项目28、29和31中任一项所述的树脂组合物,其中,将全部二胺(不包括前述通式(5)或(10)所示的化合物)设为100mol%时,前述通式(3)或(8)所示的化合物多于50mol%。
[0216]
《38》根据项目28~37中任一项所述的树脂组合物,其中,将前述树脂加热而得到的聚酰亚胺树脂膜被用于柔性基板。
[0217]
《39》根据项目28~38中任一项所述的树脂组合物,其中,将前述树脂固化而得到的聚酰亚胺树脂膜被用于柔性显示器。
[0218]
《40》一种树脂组合物的制造方法,其包括:
[0219]
使选自下述通式(3)所示的化合物、3,3
’‑
二氨基二苯基砜(33das)和4,4
’‑
二氨基二苯基砜(44das)中的至少一种二胺与酸二酐与下述通式(5)所示的含硅化合物与其它化合物发生缩聚反应,提供聚酰亚胺前体和/或聚酰亚胺。
[0220][0221]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0222]
《41》一种树脂组合物的制造方法,其包括:
[0223]
使二胺(其中不包括2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda))与酸二
酐发生缩聚反应而得到聚酰亚胺后,
[0224]
使下述通式(5)所示的含硅化合物与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物。
[0225][0226]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0227]
《42》一种树脂组合物的制造方法,其包括:
[0228]
使选自下述通式(3)所示的化合物、3,3
’‑
二氨基二苯基砜(33as)和4,4
’‑
二氨基二苯基砜(44das)中的至少一种二胺与酸二酐与其它化合物发生缩聚反应而得到聚酰亚胺后,
[0229]
使下述通式(5)所示的含硅化合物与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物。
[0230][0231]
{式中,r1各自独立地为单键或碳原子数1~10s的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0232]
《43》一种聚酰亚胺树脂膜的制造方法,其包括:
[0233]
在支承体的表面上涂布项目28~39中任一项所述的树脂组合物或通过项目40~42中任一项所述的方法而得到的树脂组合物的涂布工序;
[0234]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及
[0235]
将该聚酰亚胺树脂膜从该支承体上剥离的剥离工序。
[0236]
《44》根据项目43所述的聚酰亚胺树脂膜的制造方法,其包括:
[0237]
在前述剥离工序之前,从前述支承体侧对前述树脂组合物照射激光的照射工序。
[0238]
《45》一种显示器的制造方法,其包括:
[0239]
在支承体的表面上涂布项目28~39中任一项所述的树脂组合物或通过项目40~42中任一项所述的方法而得到的树脂组合物的涂布工序;
[0240]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;
[0241]
在该聚酰亚胺树脂膜上形成元件的元件形成工序;以及
[0242]
将形成有该元件的该聚酰亚胺树脂膜从该支承体上剥离的剥离工序。
[0243]
《46》一种层叠体的制造方法,其包括:
[0244]
在支承体的表面上涂布项目28~39中任一项所述的树脂组合物或通过项目40~42中任一项所述的方法而得到的树脂组合物的涂布工序;
[0245]
将该树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及
[0246]
在该聚酰亚胺树脂膜上形成元件的元件形成工序。
[0247]
《47》根据项目46所述的层叠体的制造方法,其还包括:
[0248]
将形成有前述元件的前述聚酰亚胺树脂膜从前述支承体上剥离的工序。
[0249]
《48》一种柔性设备的制造方法,其包括:
[0250]
通过项目46或47所述的方法来制造层叠体。
[0251]
发明的效果
[0252]
根据本发明,首先将daba、cpoda和其它含硅化合物等用作单体,可提供在显示器用途中寻求的特性优异的聚酰亚胺前体或聚酰亚胺树脂组合物。
[0253]
根据本发明,其次将daba、含硅化合物和其它化合物等用作单体,或者将daba、含硅化合物和其它化合物等用作单体,将3,3
’‑
二氨基二苯基砜(33das)和/或4,4
’‑
二氨基二苯基砜(44das)与含硅化合物与其它化合物等用作单体,可提供在显示器用途中寻求的特性优异的聚酰亚胺前体或聚酰亚胺树脂组合物。
[0254]
需要说明的是,不能视为上述记载公开了本发明的全部实施方式和本发明涉及的全部优点。本发明的进一步的实施方式及其优点可通过参照以下的记载而得以明确。
附图说明
[0255]
图1是示出作为本实施方式的显示器的例子的顶部发光型柔性有机el显示器的、比聚酰亚胺基板更靠上部的结构的示意图。
具体实施方式
[0256]
以下,针对本发明的例示实施方式(以下简写为“本实施方式”)进行详细说明。本发明不限定于本实施方式,可以在其主旨范围内进行各种变形来实施。本技术说明书中,各数值范围的上限值与下限值可以任意组合。
[0257]
〔第一方式〕
[0258]
《树脂组合物》
[0259]
〈树脂/聚酰亚胺前体/聚酰亚胺〉
[0260]
本实施方式的树脂组合物包含下述通式(6)和(7)所示结构单元的树脂,也可以包含部分进行了酰亚胺化的聚酰亚胺前体,
[0261][0262]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0263][0264]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0265]
p5或p6包含源自下述通式(10)所示含硅化合物的结构单元。
[0266][0267]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0268]
在这种部分进行了酰亚胺化的聚酰亚胺前体的情况下,与全聚酰亚胺前体相比,组合物的粘度稳定性优异,若与全聚酰亚胺相比,则从聚酰亚胺(聚酰亚胺前体)的合成容易度的观点出发是优异的。
[0269]
以前述树脂的总质量作为基准,前述含硅化合物的比例为25质量%以下。从清漆的粘度保存稳定性、过滤性的观点出发,全部树脂中的含硅化合物的比例优选为20质量%以下,更优选为10质量%以下,特别优选为5质量%以下。像这样,树脂中的含硅化合物的比例越小则清漆的粘度保存稳定性、过滤性越良好的机理尚不确定,可以认为其与已解离的含硅化合物的聚集有关。
[0270]
另一方面,从所得聚酰亚胺薄膜的残留应力的观点出发,前述含硅化合物的比例优选为5质量%以上,更优选为10质量%以上,特别优选为15质量%以上。
[0271]
全部树脂中的含硅化合物的适当比例还因所使用的二胺单体/酸二酐单体的种类/比例而异,需要综合考虑聚酰亚胺薄膜的残留应力、清漆的粘度保存稳定性、过滤性等来决定。
[0272]
前述树脂的酰亚胺化率为50%以上。从清漆的粘度保存稳定性的观点出发,前述树脂的酰亚胺化率优选为60%以上,更优选为70%以上,进一步优选为80%以上,特别优选为90%以上。像这样,酰亚胺化率越大则清漆的粘度保存稳定性越良好的机理尚不明确,可以认为其与保存清漆时的酰胺部分发生解聚有关。
[0273]
另一方面,从清漆的吸湿白浊的观点出发,前述树脂的酰亚胺化率优选为90%以
下,更优选为80%以下,进一步优选为70%以下。像这样,酰亚胺化率越小则清漆越不易发生吸湿白浊的机理尚不明确,可以认为其与聚酰亚胺/聚酰胺的溶剂溶解性有关。
[0274]
前述树脂的适当的酰亚胺化率还因所使用的二胺单体/酸二酐单体的种类/比例而异,需要综合考虑清漆的粘度保存稳定性、清漆的吸湿白浊等来决定。
[0275]
前述树脂的二胺单体中的含硅化合物比例的、酰亚胺单元与酰胺单元之差大于0且为60以下。前述树脂的二胺单体中的含硅化合物比例的、酰亚胺单元与酰胺单元之差利用下述式来求出。
[0276]
a:酰亚胺单元的二胺中的含硅化合物比例(质量%)=在酰亚胺化工序中使用的含硅化合物/在酰亚胺化工序中使用的二胺单体(包括含硅化合物)的质量的总量
×
100
[0277]
b:酰胺单元的二胺中的含硅化合物比例(质量%)=在酰亚胺化工序中使用的含硅化合物/在酰胺化工序中使用的二胺单体(包括含硅化合物)的质量的总量
×
100
[0278]
此处,a也可以换称为“构成通式(7)中的p5的二胺之中的通式(10)的比例(质量%)”。另外,b也可以换称为“构成通式(6)中的p3的二胺之中的通式(10)的比例(质量%)”。
[0279]
并且,二胺中的含硅化合物比例的、酰亚胺单元与酰胺单元之差使用上述a、b,成为b-a。
[0280]
二胺单体中的含硅化合物比例的、酰亚胺单元与酰胺单元之差表示:在具有源自含硅化合物的结构且部分进行了酰亚胺化的聚酰亚胺前体(聚酰胺酰亚胺)树脂中,酰亚胺单元和酰胺单元的含硅化合物比例的多少,值越大,则可以说酰亚胺单元越具有源自含硅化合物的结构。该值越大,则聚酰亚胺薄膜的残留应力变得越良好,若该值小,则清漆的起泡性/过滤性/聚酰亚胺薄膜的雾度(浊度)变得良好。并且,在该值为上述范围的情况下,能够兼顾清漆的起泡性/过滤性/聚酰亚胺薄膜的残留应力/雾度(浊度)等各种特性,故而优选。
[0281]
另外,其中,前述树脂所使用的二胺为选自下述通式(8)的化合物、3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)和9,9-双(4-氨基苯基)芴(bafl)中的至少一种化合物时,清漆/聚酰亚胺的特性变得更良好,故而优选。
[0282][0283]
另外,本发明的其它实施方式所述的树脂组合物也可以包含聚酰亚胺前体,所述聚酰亚胺前体包含下述通式(6)和(7)所示的结构单元,且部分进行了酰亚胺化。
[0284][0285]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数。}
[0286][0287]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数。}
[0288]
p5或p6可以包含源自下述通式(10)所示含硅化合物的结构单元,
[0289][0290]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50。}
[0291]
并且,所述树脂组合物可以满足下述(一)或(二)中的任意者,
[0292]
(一)前述p3和/或p5包含源自下述通式(8)所示化合物的结构单元,
[0293]
或者
[0294]
(二)前述p4和/或p6包含源自下述通式(9)所示化合物的结构单元。
[0295][0296]
二胺
[0297]
树脂组合物包含选自下述通式(8)的化合物、3,3
’‑
二氨基二苯基砜(33das)、4,4
’‑
二氨基二苯基砜(44das)、9,9-双(4-氨基苯基)芴(bafl)或2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)的各种二胺化合物中的至少一种结构单元。
[0298][0299]
通式(8)所示的二胺化合物之中,从聚酰亚胺薄膜的透明性、yi的观点出发,优选为3,5-二氨基苯甲酸(daba)。通过使用选自daba、33das、44das、bafl和6foda中的至少一种作为二胺化合物,从而聚酰亚胺薄膜的机械特性提高(尤其是拉伸伸长率),能够提高耐热性,故而优选。
[0300]
全部二胺(包括上述通式的(10)中的l1和l2为氨基的化合物)中的daba的含量可以
超过50摩尔%、超过55摩尔%、或者为70摩尔%以上、或者为90摩尔%以上、或者为95摩尔%以上。daba的量越多,则聚酰亚胺薄膜的拉伸伸长率变得越大,故而优选。
[0301]
作为除通式(8)之外的二胺,可列举出对苯二胺(pda)、间苯二胺、2,2
’‑
二甲基联苯胺(mtb)、4,4
’‑
二氨基二苯硫醚、3,4
’‑
二氨基二苯硫醚、3,3
’‑
二氨基二苯硫醚、4,4
’‑
二氨基联苯、3,4
’‑
二氨基联苯、3,3
’‑
二氨基联苯、4,4
’‑
二氨基二苯甲酮、3,4
’‑
二氨基二苯甲酮、3,3
’‑
二氨基二苯甲酮、4,4
’‑
二氨基二苯基甲烷、3,4
’‑
二氨基二苯基甲烷、3,3
’‑
二氨基二苯基甲烷、1,4-双(4-氨基苯氧基)苯、1,3-双(4-氨基苯氧基)苯、1,3-双(3-氨基苯氧基)苯、双〔4-(4-氨基苯氧基)苯基〕砜、4,4-双(4-氨基苯氧基)联苯、4,4-双(3-氨基苯氧基)联苯、双〔4-(4-氨基苯氧基)苯基〕醚、双〔4-(3-氨基苯氧基)苯基〕醚、1,4-双(4-氨基苯基)苯、1,3-双(4-氨基苯基)苯、9,10-双(4-氨基苯基)蒽、2,2-双(4-氨基苯基)丙烷、2,2-双(4-氨基苯基)六氟丙烷、2,2-双〔4-(4-氨基苯氧基)苯基)丙烷、2,2-双〔4-(4-氨基苯氧基)苯基)六氟丙烷和1,4-双(3-氨基丙基二甲基甲硅烷基)苯、1,3-双[1-(4-氨基苯基)-1-甲基乙基]苯](bisam)等。
[0302]
除通式(8)之外的二胺优选为选自由1,3-双[1-(4-氨基苯基)-1-甲基乙基]苯](bisam)和1,4-环己烷二胺(chda)组成的组中的至少1种。
[0303]
酸二酐
[0304]
树脂组合物包含选自下述通式(9)所示的化合物(也称为cpoda)、4,4
’‑
氧双邻苯二甲酸酐(odpa)或9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)的各种酸二酐化合物中的至少一种结构单元。
[0305][0306]
若具有这些结构单元,则能够提高所得聚酰亚胺薄膜的透明性、yi、耐热性的机械特性,故而优选。
[0307]
全部酸二酐(包括上述通式(10)中的l1和l2为酸酐基的化合物)中的cpoda、odpa和bpaf的含量可以为50摩尔%以上、60摩尔%以上、或者70摩尔%以上、或者90摩尔%以上、或者95摩尔%以上。cpoda、odpa和bpaf的量越多,则聚酰亚胺薄膜的透明性越会提高,故而优选。
[0308]
作为除通式(9)之外的酸二酐,可列举出均苯四甲酸二酐(pmda)、3,3’,4,4
’‑
联苯四羧酸二酐(bpda)、2,2’,3,3
’‑
联苯四羧酸二酐、4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐(6fda)、5-(2,5-二氧四氢-3-呋喃基)-3-甲基-环己烯-1,2二羧酸酐、1,2,3,4-苯四羧酸二酐、3,3’,4,4
’‑
二苯甲酮四羧酸二酐、2,2’,3,3
’‑
二苯甲酮四羧酸二酐、3,3’,4,4
’‑
二苯基砜四羧酸二酐、亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,1-亚乙基-4,4
’‑
双邻苯二甲酸二酐、2,2-亚丙基-4,4
’‑
双邻苯二甲酸二酐、1,2-亚乙基-4,4
’‑
双邻苯二甲酸二酐、1,3-三亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,4-四亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,5-五亚甲基-4,4
’‑
双邻苯二甲酸二酐、4,4
’‑
氧双邻苯二甲酸二酐、对亚苯基双偏苯三酸酐、硫代-4,4
’‑
双邻苯二甲酸二酐、磺酰基-4,4
’‑
双邻苯二甲酸二酐、1,3-双(3,4-二羧基苯基)苯二酐、1,3-双(3,4-二羧基苯氧基)苯二酐、1,4-双(3,4-二羧基苯氧基)苯二酐、1,3-双[2-(3,4-二羧基
苯基)-2-丙基]苯二酐、1,4-双[2-(3,4-二羧基苯基)-2-丙基]苯二酐、双[3-(3,4-二羧基苯氧基)苯基]甲烷二酐、双[4-(3,4-二羧基苯氧基)苯基]甲烷二酐、2,2-双[3-(3,4-二羧基苯氧基)苯基]丙烷二酐、2,2-双[4-(3,4-二羧基苯氧基)苯基]丙烷二酐、双(3,4-二羧基苯氧基)二甲基硅烷二酐、1,3-双(3,4-二羧基苯基)-1,1,3,3-四甲基二硅氧烷二酐、2,3,6,7-萘四羧酸二酐、1,4,5,8-萘四羧酸二酐、1,2,5,6-萘四羧酸二酐、3,4,9,10-苝四羧酸二酐、2,3,6,7-蒽四羧酸二酐和1,2,7,8-菲四羧酸二酐、1,2,4,5-环己烷四羧酸二酐(hpmda)和1,2,3,4-环丁烷四羧酸二酐(cbda)、下述结构的化合物(bzda);
[0309][0310]
下述结构的化合物(bnbda)等。
[0311][0312]
除上述通式(9)之外的酸二酐优选为选自由bzda、bnbda、1,2,4,5-环己烷四羧酸二酐(hpmda)组成的组中的至少1种。酸二酐可以单独使用一种,也可以组合使用两种以上。
[0313]
〈含硅化合物〉
[0314]
本实施方式中的聚酰亚胺前体可以一同具有上述式(6)所示的结构和上述式(7)所示的结构。以聚酰亚胺前体的质量作为基准,聚酰亚胺前体中的源自含硅化合物的结构的含有比例优选为5质量%以上且40质量%以下。若聚酰亚胺前体在该数值范围内包含源自含硅化合物的结构,则能够在所得聚酰亚胺薄膜中兼顾低残留应力和高度的透明性、耐热性,故而优选。以聚酰亚胺前体的质量作为基准,源自含硅化合物的结构的含有比例可以为6质量%以上或7质量%以上,另外,可以为30质量%以下或25质量%以下。
[0315]
本实施方式中的聚酰亚胺/聚酰亚胺前体具有源自含硅化合物的结构。因此,在本实施方式中的聚酰亚胺前体的合成中使用的含硅化合物可以是具有能够与四羧酸二酐和二胺之中的至少一者发生共缩合的反应性基团的化合物。
[0316]
这种含硅化合物可列举出例如下述式(10)所示的化合物。
[0317][0318]
{式中,
[0319]
r1各自独立地为单键或碳原子数1~10的二价有机基团,
[0320]
r2和r3各自独立地为碳原子数1~10的一价有机基团,r2和r3之中的至少一者为碳原子数1~5的一价脂肪族烃基,
[0321]
r4和r5各自独立地为碳原子数1~10的一价有机基团,r4和r5之中的至少一者为碳原子数6~10的一价芳香族基团,
[0322]
r6和r7各自独立地为碳原子数1~10的一价有机基团,r6和r7之中的至少一者为包含碳原子数2~10的不饱和脂肪族烃基的有机基团,
[0323]
l1和l2各自独立地为包含酸酐结构的一价有机基团、氨基、异氰酸酯基、羧基、烷氧基羰基、卤代羰基、羟基、环氧基或巯基,
[0324]
i和j各自独立地为1~200的整数,
[0325]
k为0~200的整数,并且
[0326]
满足0.05≤j/(i+j+k)≤0.50的关系。}
[0327]
式(10)中的r1各自独立地为单键或碳原子数1~10的二价有机基团。碳原子数1~10的二价有机基团可以为直链状、环状和支链状中的任意者,可以饱和也可以不饱和。作为碳原子数1~10的二价脂肪族烃基,可列举出例如亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚仲丁基、亚叔丁基、亚正戊基、亚新戊基、亚正己基、亚正庚基、亚正辛基、亚正壬基、亚正癸基等直链或支链亚烷基;亚环丙基、亚环丁基、亚环戊基、亚环己基、亚环庚基、亚环辛基等亚环烷基。作为碳原子数1~10的二价脂肪族烃基,优选为选自由亚乙基、亚正丙基和亚异丙基组成的组中的至少1种。
[0328]
式(10)中的r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基。
[0329]
碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~10的一价有机基团,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。
[0330]
碳原子数1~5的一价脂肪族烃基可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~5的一价脂肪族烃基,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基等直链或支链烷基;环丙基、环丁基、环戊基等环烷基等。作为碳原子数1~5的一价脂肪族烃基,优选为选自由甲基、乙基和正丙基组成的组中的至少1种。
[0331]
式(10)中的r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团。碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~10的一价有机基团,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。作为碳原子数6~10的一价芳香族基团,可列举出例如苯基、甲苯基、二甲苯基、α-萘基、β-萘基等,优选为苯基、甲苯基或二甲苯基。
[0332]
式(10)中的r6和r7各自独立地为碳原子数1~10的一价有机基团,至少一者为具有不饱和脂肪族烃基的有机基团。碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者。作为碳原子数1~10的一价有机基团,可列举出例如甲基、乙基、正丙基、异
丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。作为碳原子数1~10的一价有机基团,优选为选自由甲基、乙基和苯基组成的组中的至少1种。
[0333]
具有不饱和脂肪族烃基的有机基团可以为碳原子数3~10的不饱和脂肪族烃基,可以为直链状、环状、支链状中的任意者。作为碳原子数3~10的不饱和脂肪族烃基,可列举出例如乙烯基、烯丙基、1-丙烯基、3-丁烯基、2-丁烯基、戊烯基、环戊烯基、己烯基、环己烯基、庚烯基、辛烯基、壬烯基、癸烯基、乙炔基、丙炔基、丁炔基、戊炔基、己炔基等。作为碳原子数3~10的不饱和脂肪族烃基,优选为选自由乙烯基、烯丙基和3-丁烯基组成的组中的至少1种。
[0334]
式(10)中的r1~r7的一部分或全部氢原子任选被f、cl、br等卤素原子等取代基取代,也可以未取代。
[0335]
式(10)中的l1和l2各自独立地为包含酸酐结构的一价有机基团(也称为酸酐基)、氨基、异氰酸酯基、羧基、烷氧基羰基、卤代羰基、羟基、环氧基或巯基。
[0336]
作为包含酸酐结构的一价有机基团,可列举出例如下述式所示的2,5-二氧四氢呋喃-3-基。
[0337][0338]
{上述式中,“*”表示连接键。}
[0339]
这些之中,优选为氨基、酸酐基,从树脂组合物的粘度稳定性的观点出发,更优选为氨基。
[0340]
烷氧基羰基中的烷氧基可以为碳原子数1~6的烷氧基,可以为例如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基等。
[0341]
卤代羰基中的卤素原子优选为除氟原子之外的卤素原子,更优选为氯原子或碘原子。
[0342]
从树脂组合物的过滤性的观点出发,式(10)所示的含硅化合物的官能团当量优选为800以上,更优选为1000以上,进一步优选为1500以上。另一方面,在官能团当量为500以下的情况下,过滤性有时变差。此处,官能团当量是每1mol官能团中的含硅化合物的分子量(单位:g/mol)。官能团当量可利用公知方法进行测定。另外,在含硅化合物的官能团当量为800以上的情况下,聚酰亚胺薄膜在氮气气氛下的残留应力小,故而优选。作为其理由,可认为这是因为:在官能团当量为特定值以上的情况下,有机硅区域(domain)增加,应力得以缓和。
[0343]
需要说明的是,官能团当量可按照已有的标准等进行测定。
[0344]
式(10)中的i为1~200的整数、优选为2~100的整数、更优选为4~80的整数、进一步优选为8~40的整数。j和k各自独立地为0~200的整数、优选为0~50的整数、更优选为0~20的整数、进一步优选为0~50的整数。
[0345]
若树脂组合物中的聚酰亚胺具有源自式(10)的结构,则聚酰亚胺薄膜在氮气气氛
下测得的残留应力良好(小),故而优选。作为在氮气气氛下进行测定的原因,是因为:在显示器的工艺中,在聚酰亚胺薄膜上形成sio、sin等的无机膜时,有时暴露在氮气气氛下,寻求在氮气气氛下的残留应力小。
[0346]
〔第二方式〕
[0347]
《树脂组合物》
[0348]
〈树脂/聚酰亚胺前体/聚酰亚胺〉
[0349]
本实施方式的树脂组合物包含下述通式(1)或(2)所示结构单元的树脂,也可以包含聚酰亚胺前体(以下也称为全聚酰亚胺前体)或聚酰亚胺(以下也称为全聚酰亚胺)。
[0350][0351]
{式中,p1表示二价有机基团;p2表示四价有机基团;并且,p表示正整数。}
[0352][0353]
{式中,p1表示二价有机基团;p2表示四价有机基团;并且,p表示正整数。}
[0354]
p1包含源自下述通式(3)所示化合物的结构单元,并且,根据期望,p2可以包含源自下述通式(4)所示化合物的结构单元。
[0355][0356][0357]
另外,p1或p2包含源自下述通式(5)所示含硅化合物的结构单元。
[0358][0359]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50;且官能团当量为8000以上。}
[0360]
聚酰亚胺通过对聚酰亚胺前体进行热酰亚胺化来获得,也可以进行化学酰亚胺化。从所得聚酰亚胺薄膜的透明性的观点出发,优选为热酰亚胺化。
[0361]
另外,从组合物的粘度稳定性的观点出发,聚酰亚胺树脂组合物与聚酰亚胺前体树脂组合物相比更为优选。
[0362]
另外,本发明的其它实施方式所述的树脂组合物也可以包含聚酰亚胺前体,所述聚酰亚胺前体包含下述通式(6)和(7)所示的结构单元,且部分进行了酰亚胺化。
[0363][0364]
{式中,p3表示二价有机基团;p4表示四价有机基团;并且,p表示正整数;p3不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)或源自其的结构单元。}
[0365][0366]
{式中,p5表示二价有机基团;p6表示四价有机基团;并且,q表示正整数;p5不含2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚(6foda)或源自其的结构单元。}
[0367]
p3和/或p5也可以包含源自下述通式(8)所示化合物的结构单元。
[0368][0369]
另外,p3和/或p5可以包含至少一个源自3,3
’‑
二氨基二苯基砜(33das)或4,4
’‑
二氨基二苯基砜(44das)中的各种化合物的结构单元。
[0370]
另外,p3和/或p5可以包含至少一个源自9,9-双(4-氨基苯基)芴(bafl)或下述通式的化合物(bisam)中的各种化合物的结构单元。
[0371][0372]
这些之中,从聚酰亚胺薄膜的伸长率、机械特性的观点出发,优选为前述下述通式(8)的化合物、3,3
’‑
二氨基二苯基砜(33das)和/或4,4
’‑
二氨基二苯基砜(44das)。另一方面,从清漆的粘度稳定性、过滤性的观点、在酰亚胺化时产生的异物的观点出发,前述p3和/或p5不优选使用6foda。
[0373]
p4和/或p6也可以包含源自下述通式(9)所示化合物的结构单元。
[0374][0375]
另外,p4和/或p6可以包含至少一个源自:
[0376]
4,4
’‑
氧双邻苯二甲酸酐(odpa)、
[0377]
4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐(6fda)、
[0378]
9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)、
[0379]
下述通式的化合物(bzda)、
[0380][0381]
以及下述通式的化合物(bnbda)、
[0382][0383]
中的化合物的结构单元。
[0384]
这些之中,从清漆的粘度稳定性、过滤性的观点、聚酰亚胺薄膜的透明性的观点出发,优选为通式(9)的化合物、odpa。
[0385]
另外,一个实施方式中,p3或p4包含源自下述通式(10)所示含硅化合物的结构单元。
[0386][0387]
{式中,r1各自独立地为单键或碳原子数1~10的二价有机基团;r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基;r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团;r6和r7各自独立地为碳原子数1~10的一价有机基团;l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基;i为1~200的整数;j和k各自独立地为0~200的整数;0≤j/(i+j+k)≤0.50;且官能团当量为8000以上。}
[0388]
在这种部分进行了酰亚胺化的聚酰亚胺前体的情况下,与前述全聚酰亚胺前体相比,组合物的粘度稳定性优异,若与前述全聚酰亚胺相比,则从聚酰亚胺(聚酰亚胺前体)的合成容易度的观点出发是优异的。
[0389]
另外,通式(10)中,l1和l2各自独立地为氨基、酸酐基、异氰酸酯基、羧基、酸酯基、酰卤基、羟基、环氧基或巯基,它们之中,从树脂特性或薄膜特性的观点出发,优选为氨基。
[0390]
二胺
[0391]
树脂组合物包含源自下述通式(3)所示二胺化合物的结构单元。
[0392][0393]
通式(3)所示的二胺化合物之中,从聚酰亚胺薄膜的透明性、yi的观点出发,优选为3,5-二氨基苯甲酸(daba)。若具有该结构单元,则所得聚酰亚胺薄膜的机械特性提高(尤其是拉伸伸长率),能够提高耐热性,故而优选。这种特性提高的理由尚不明确,可认为是因为:通过羧基的作用而发挥出分子间相互作用。
[0394]
全部二胺(包括上述通式的(5)、(10)中的l1和l2为氨基的化合物)中的daba的含量可以超过50摩尔%、超过55摩尔%、或者为70摩尔%以上、或者为90摩尔%以上、或者为95摩尔%以上。daba的量越多,则聚酰亚胺薄膜的拉伸伸长率变得越大,故而优选。
[0395]
另外,树脂组合物包含源自33das和/或44das的结构单元。从兼顾聚酰亚胺薄膜的厚度方向rth(延迟)和机械特性的观点出发,33das、44das优选使用33das与44das的混合物。
[0396]
全部二胺(包括上述通式的(5)、(10)中的l1和l2为氨基的化合物)中的33das和/或44das的总含量可以超过50摩尔%、超过55摩尔%、或者为70摩尔%以上、或者为90摩尔%以上、或者为95摩尔%以上。33das和/或44das的量越多,则聚酰亚胺薄膜的机械强度越会提高,故而优选。
[0397]
作为除通式(3)之外的二胺,可列举出对苯二胺(pda)、间苯二胺、2,2
’‑
二甲基联苯胺(mtb)、4,4
’‑
二氨基二苯硫醚、3,4
’‑
二氨基二苯硫醚、3,3
’‑
二氨基二苯硫醚、4,4
’‑
二氨基联苯、3,4
’‑
二氨基联苯、3,3
’‑
二氨基联苯、4,4
’‑
二氨基二苯甲酮、3,4
’‑
二氨基二苯甲酮、3,3
’‑
二氨基二苯甲酮、4,4
’‑
二氨基二苯基甲烷、3,4
’‑
二氨基二苯基甲烷、3,3
’‑
二氨基二苯基甲烷、1,4-双(4-氨基苯氧基)苯、1,3-双(4-氨基苯氧基)苯、1,3-双(3-氨基苯氧基)苯、双〔4-(4-氨基苯氧基)苯基〕砜、4,4-双(4-氨基苯氧基)联苯、4,4-双(3-氨基苯氧基)联苯、双〔4-(4-氨基苯氧基)苯基〕醚、双〔4-(3-氨基苯氧基)苯基〕醚、1,4-双(4-氨基苯基)苯、1,3-双(4-氨基苯基)苯、9,10-双(4-氨基苯基)蒽、2,2-双(4-氨基苯基)丙烷、2,2-双(4-氨基苯基)六氟丙烷、2,2-双〔4-(4-氨基苯氧基)苯基)丙烷、2,2-双〔4-(4-氨基苯氧基)苯基)六氟丙烷和1,4-双(3-氨基丙基二甲基甲硅烷基)苯、9,9-双(4-氨基苯基)芴(bafl)、1,3-双[1-(4-氨基苯基)-1-甲基乙基]苯](bisam)等。
[0398]
这些之中,优选为选自由9,9-双(4-氨基苯基)芴(bafl)、1,3-双[1-(4-氨基苯基)-1-甲基乙基]苯](bisam)和1,4-环己烷二胺(chda)组成的组中的至少1种。
[0399]
从所得聚酰亚胺树脂膜的耐热性和机械强度的观点出发,除通式(3)33das和/或44das之外的二胺更优选为选自由9,9-双(4-氨基苯基)芴(bafl)、1,3-双[1-(4-氨基苯基)-1-甲基乙基]苯](bisam)组成的组中的至少1种以上。
[0400]
酸二酐
[0401]
树脂组合物包含源自下述通式(4)所示化合物(也称为cpoda)的结构单元。
[0402][0403]
若具有该结构单元,则能够提高所得聚酰亚胺薄膜的透明性、yi、耐热性的机械特性,故而优选。
[0404]
全部酸二酐(包括上述通式的(5)、(10)中的l1和l2为酸酐基的化合物)中的cpoda的含量可以为50摩尔%以上、60摩尔%以上、或者为70摩尔%以上、或者为90摩尔%以上、或者为95摩尔%以上。cpoda的量越多,则聚酰亚胺薄膜的透明性越会提高,故而优选。
[0405]
另外,树脂组合物包含源自odpa的结构单元。若具有该结构单元,则从清漆的粘度稳定性、过滤性的观点出发,能够提高所得聚酰亚胺薄膜的透明性,故而优选。
[0406]
全部酸二酐(包括上述通式的(5)、(10)中的l1和l2为酸酐基的化合物)中的odpa的含量可以为50摩尔%以上、60摩尔%以上、或者为70摩尔%以上、或者为90摩尔%以上、或者为95摩尔%以上。odpa的量越多,则聚酰亚胺薄膜的透明性越会提高,故而优选。
[0407]
作为除通式(4)之外的酸二酐,可列举出均苯四甲酸二酐(pmda)、3,3’,4,4
’‑
联苯四羧酸二酐(bpda)、2,2’,3,3
’‑
联苯四羧酸二酐、4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐(6fda)、5-(2,5-二氧四氢-3-呋喃基)-3-甲基-环己烯-1,2二羧酸酐、1,2,3,4-苯四羧酸二酐、3,3’,4,4
’‑
二苯甲酮四羧酸二酐、2,2’,3,3
’‑
二苯甲酮四羧酸二酐、3,3’,4,4
’‑
二苯基砜四羧酸二酐、亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,1-亚乙基-4,4
’‑
双邻苯二甲酸二酐、2,2-亚丙基-4,4
’‑
双邻苯二甲酸二酐、1,2-亚乙基-4,4
’‑
双邻苯二甲酸二酐、1,3-三亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,4-四亚甲基-4,4
’‑
双邻苯二甲酸二酐、1,5-五亚甲基-4,4
’‑
双邻苯二甲酸二酐、4,4
’‑
氧双邻苯二甲酸二酐、对亚苯基双偏苯三酸酐、硫代-4,4
’‑
双邻苯二甲酸二酐、磺酰基-4,4
’‑
双邻苯二甲酸二酐、1,3-双(3,4-二羧基苯基)苯二酐、1,3-双(3,4-二羧基苯氧基)苯二酐、1,4-双(3,4-二羧基苯氧基)苯二酐、1,3-双[2-(3,4-二羧基苯基)-2-丙基]苯二酐、1,4-双[2-(3,4-二羧基苯基)-2-丙基]苯二酐、双[3-(3,4-二羧基苯氧基)苯基]甲烷二酐、双[4-(3,4-二羧基苯氧基)苯基]甲烷二酐、2,2-双[3-(3,4-二羧基苯氧基)苯基]丙烷二酐、2,2-双[4-(3,4-二羧基苯氧基)苯基]丙烷二酐、双(3,4-二羧基苯氧基)二甲基硅烷二酐、1,3-双(3,4-二羧基苯基)-1,1,3,3-四甲基二硅氧烷二酐、2,3,6,7-萘四羧酸二酐、1,4,5,8-萘四羧酸二酐、1,2,5,6-萘四羧酸二酐、3,4,9,10-苝四羧酸二酐、2,3,6,7-蒽四羧酸二酐和1,2,7,8-菲四羧酸二酐、9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)、4,4
’‑
氧双邻苯二甲酸酐(odpa)、1,2,4,5-环己烷四羧酸二酐(hpmda)和1,2,3,4-环丁烷四羧酸二酐(cbda)、下述结构的化合物(bzda);
[0408][0409]
下述结构的化合物(bnbda)等。
[0410][0411]
除上述通式(4)之外的酸二酐优选为选自由6fda、9,9-双(3,4-二羧基苯基)芴二酸酐(bpaf)、bzda、bnbda、1,2,4,5-环己烷四羧酸二酐(hpmda)组成的组中的至少1种。酸二酐可以单独使用一种,也可以组合使用两种以上。
[0412]
〈含硅化合物〉
[0413]
本实施方式中的聚酰亚胺前体可以一同具有上述式(1)所示的结构和上述式(2)所示的结构。以聚酰亚胺前体的质量作为基准,聚酰亚胺前体中的源自含硅化合物的结构的含有比例优选为5质量%以上且40质量%以下。若聚酰亚胺前体在该数值范围内包含源自含硅化合物的结构,则能够在所得聚酰亚胺薄膜中兼顾低残留应力和高度的透明性、耐热性,故而优选。以聚酰亚胺前体的质量作为基准,源自含硅化合物的结构的含有比例可以为6质量%以上或7质量%以上,另外,可以为30质量%以下或25质量%以下。
[0414]
本实施方式中的聚酰亚胺/聚酰亚胺前体具有源自含硅化合物的结构。因此,在本实施方式中的聚酰亚胺前体的合成中使用的含硅化合物可以是具有能够与四羧酸二酐和二胺之中的至少一者发生共缩合的反应性基团的化合物。
[0415]
这种含硅化合物可列举出例如下述式(5)所示的化合物。
[0416][0417]
{式中,
[0418]
r1各自独立地为单键或碳原子数1~10的二价有机基团,
[0419]
r2和r3各自独立地为碳原子数1~10的一价有机基团,r2和r3之中的至少一者为碳原子数1~5的一价脂肪族烃基,
[0420]
r4和r5各自独立地为碳原子数1~10的一价有机基团,r4和r5之中的至少一者为碳原子数6~10的一价芳香族基团,
[0421]
r6和r7各自独立地为碳原子数1~10的一价有机基团,r6和r7之中的至少一者为包含碳原子数2~10的不饱和脂肪族烃基的有机基团,
[0422]
l1和l2各自独立地为包含酸酐结构的一价有机基团、氨基、异氰酸酯基、羧基、烷氧基羰基、卤代羰基、羟基、环氧基或巯基,
[0423]
i和j各自独立地为1~200的整数,
[0424]
k为0~200的整数,并且
[0425]
满足0.05≤j/(i+j+k)≤0.50的关系。}
[0426]
式(5)中的r1各自独立地为单键或碳原子数1~10的二价有机基团。碳原子数1~10的二价有机基团可以为直链状、环状和支链状中的任意者,可以饱和也可以不饱和。作为碳原子数1~10的二价脂肪族烃基,可列举出例如亚甲基、亚乙基、亚正丙基、亚异丙基、亚正丁基、亚仲丁基、亚叔丁基、亚正戊基、亚辛戊基、亚正己基、亚正庚基、亚正辛基、亚正壬
基、亚正癸基等直链或支链亚烷基;亚环丙基、亚环丁基、亚环戊基、亚环己基、亚环庚基、亚环辛基等亚环烷基。作为碳原子数1~10的二价脂肪族烃基,优选为选自由亚乙基、亚正丙基和亚异丙基组成的组中的至少1种。
[0427]
式(5)中的r2和r3各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数1~5的一价脂肪族烃基。
[0428]
碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~10的一价有机基团,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。
[0429]
碳原子数1~5的一价脂肪族烃基可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~5的一价脂肪族烃基,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基等直链或支链烷基;环丙基、环丁基、环戊基等环烷基等。作为碳原子数1~5的一价脂肪族烃基,优选为选自由甲基、乙基和正丙基组成的组中的至少1种。
[0430]
式(5)中的r4和r5各自独立地为碳原子数1~10的一价有机基团,至少一者为碳原子数6~10的一价芳香族基团。碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者,可以饱和也可以不饱和。例如,作为碳原子数1~10的一价有机基团,可列举出甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。作为碳原子数6~10的一价芳香族基团,可列举出例如苯基、甲苯基、二甲苯基、α-萘基、β-萘基等,优选为苯基、甲苯基或二甲苯基。
[0431]
式(5)中的r6和r7各自独立地为碳原子数1~10的一价有机基团,至少一者为具有不饱和脂肪族烃基的有机基团。碳原子数1~10的一价有机基团可以为直链状、环状、支链状中的任意者。作为碳原子数1~10的一价有机基团,可列举出例如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、叔丁基、正戊基、新戊基、正己基、正庚基、正辛基、正壬基、正癸基等直链或支链烷基;环丙基、环丁基、环戊基、环己基、环庚基、环辛基等环烷基;苯基、甲苯基、二甲苯基、α-萘基、β-萘基等芳香族基团等。作为碳原子数1~10的一价有机基团,优选为选自由甲基、乙基和苯基组成的组中的至少1种。
[0432]
具有不饱和脂肪族烃基的有机基团可以为碳原子数3~10的不饱和脂肪族烃基,可以为直链状、环状、支链状中的任意者。作为碳原子数3~10的不饱和脂肪族烃基,可列举出例如乙烯基、烯丙基、1-丙烯基、3-丁烯基、2-丁烯基、戊烯基、环戊烯基、己烯基、环己烯基、庚烯基、辛烯基、壬烯基、癸烯基、乙炔基、丙炔基、丁炔基、戊炔基、己炔基等。作为碳原子数3~10的不饱和脂肪族烃基,优选为选自由乙烯基、烯丙基和3-丁烯基组成的组中的至少1种。
[0433]
式(5)中的r1~r7的一部分或全部氢原子任选被f、cl、br等卤素原子等取代基取代,也可以未取代。
[0434]
式(5)中的l1和l2各自独立地为包含酸酐结构的一价有机基团(也称为酸酐基)、氨
基、异氰酸酯基、羧基、烷氧基羰基、卤代羰基、羟基、环氧基或巯基。它们之中,作为l1和l2,从树脂特性或薄膜特性的观点出发,优选为氨基。
[0435]
作为包含酸酐结构的一价有机基团,可列举出例如下述式所示的2,5-二氧四氢呋喃-3-基。
[0436][0437]
{上述式中,“*”表示连接键。}
[0438]
这些之中,优选为氨基、酸酐基,从树脂组合物的粘度稳定性的观点出发,更优选为氨基。
[0439]
烷氧基羰基中的烷氧基可以为碳原子数1~6的烷氧基,可以为例如甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、叔丁氧基等。
[0440]
卤代羰基中的卤素原子优选为除氟原子之外的卤素原子,更优选为氯原子或碘原子。
[0441]
从树脂组合物的过滤性的观点出发,式(5)所示的含硅化合物的官能团当量优选为800以上,更优选为1000以上,进一步优选为1500以上。另一方面,在官能团当量为500以下的情况下,过滤性有时变差。此处,官能团当量是每1mol官能团中的含硅化合物的分子量(单位:g/mol)。官能团当量可利用公知方法进行测定。另外,在含硅化合物的官能团当量为800以上的情况下,聚酰亚胺薄膜在氮气气氛下的残留应力小,故而优选。作为其理由,可以认为这是因为:在官能团当量为特定值以上的情况下,有机硅区域(domain)增加,应力得以缓和。
[0442]
需要说明的是,官能团当量可按照已有的标准等进行测定。
[0443]
式(5)中的i为1~200的整数、优选为2~100的整数、更优选为4~80的整数、进一步优选为8~40的整数。j和k各自独立地为0~200的整数、优选为0~50的整数、更优选为0~20的整数、进一步优选为0~50的整数。
[0444]
若树脂组合物中的聚酰亚胺具有源自式(5)的结构,则聚酰亚胺薄膜在氮气气氛下测得的残留应力良好(小),故而优选。作为在氮气气氛下进行测定的原因,是因为:在显示器的工艺中,在聚酰亚胺薄膜上形成sio、sin等的无机膜时,有时暴露在氮气气氛下,寻求在氮气气氛下的残留应力小。
[0445]
〔第一方式和第二方式中共通的构成要素和优选实施方式〕
[0446]
以下,针对本发明的第一方式和第二方式中共通的构成要素和本发明的优选实施方式进行说明。需要说明的是,第一方式与第二方式的构成要素可以互换或组合。
[0447]
〈含硅二胺〉
[0448]
从单体的种类、成本的观点以及所得聚酰亚胺前体的分子量的观点出发,上述说明的通式(5)或(10)的含硅化合物优选为含硅二胺。作为含硅二胺,优选为例如下述式(11)所示的二氨基(聚)硅氧烷。
[0449][0450]
{式中,p5各自独立地表示二价烃基,任选相同或不同,p3和p4与通式(5)或(10)中定义的r2、r3相同,l表示1~200的整数。}
[0451]
作为上述通式(11)中的p3和p4的优选结构,可列举出甲基、乙基、丙基、丁基和苯基等。这些之中,优选为甲基。
[0452]
上述通式(11)中的l为1~200的整数,从使用式(11)所示的含硅二胺而得到的聚酰亚胺的耐热性的观点出发,优选为3~200的整数。
[0453]
通式(11)所示的化合物的官能团当量的优选范围与前述通式(10)所示的含硅化合物相同。
[0454]
含硅二胺的共聚比例相对于聚酰亚胺前体/聚酰亚胺的总质量优选为0.5~30质量%、更优选为1.0质量%~25质量%、进一步优选为1.5质量%~20质量%。在含硅二胺为0.5质量%以上的情况下,能够有效地降低与支承体之间产生的残留应力。在含硅二胺为30质量%以下的情况下,所得聚酰亚胺薄膜的透明性(尤其是低雾度)良好,从高的总光线透射率的实现和高的玻璃化转变温度的观点出发是优选的。
[0455]
作为在聚酰亚胺前体/聚酰亚胺中使用的单体的含硅化合物如上所述,可以使用申请时的技术常识来合成,也可以使用市售品。作为市售品,可列举出两末端胺改性甲基苯基硅油(信越化学公司制:x22-1660b-3(官能团当量为2200)、x22-9409(官能团当量为670))、两末端酸酐改性甲基苯基硅油(信越化学公司制:x22-168-p5-b(官能团当量为2100))、两末端环氧改性甲基苯基硅油(信越化学公司制:x22-2000(官能团当量为620))、两末端氨基改性二甲基有机硅(信越化学公司制:pam-e(官能团当量为130)、x22-161a(官能团当量为800)、x22-161b(官能团当量为1500)、kf8012(官能团当量为2200)、东丽道康宁公司制的by16-853u(官能团当量为450)、jnc公司制的silaplane fm3311(数均分子量为1000))、两末端环氧改性二甲基有机硅(信越化学公司制:x-22-163a(官能团当量为1750)、两末端脂环式环氧改性二甲基有机硅(信越化学公司制:x-22-169b(官能团当量为1700))、两末端羟基改性二甲基有机硅(信越化学公司制:kf-6000)、两末端巯基改性二甲基有机硅(信越化学公司制:x-22-167b(官能团当量为1700))、两末端酸酐改性二甲基有机硅(信越化学公司制:x-22-168a(官能团当量为1000))等。这些之中,从价格、提高耐化学药品性和提高tg的观点出发,优选为两末端胺改性二甲基硅油。
[0456]
〈溶剂〉
[0457]
典型而言,树脂组合物包含溶剂。作为溶剂,优选聚酰亚胺/聚酰亚胺前体的溶解性良好且能够适当控制树脂组合物的溶液粘度的溶剂,可以将聚酰亚胺前体的反应溶剂用作组合物的溶剂。其中,优选为n-甲基-2-吡咯烷酮(nmp)、γ-丁内酯(gbl)、第一方式所述的上述通式(9)所示的化合物、第二方式所述的上述通式(4)所示的化合物等。作为溶剂组成的具体例,可列举出n-甲基-2-吡咯烷酮(nmp)单独、或者n-甲基-2-吡咯烷酮(nmp)与γ-丁内酯(gbl)的混合溶剂等。nmp与gbl的质量比可以为例如nmp:gbl(质量比)=10:90~90:10。
[0458]
〈追加成分〉
[0459]
本实施方式的树脂组合物中,在包含聚酰亚胺/聚酰亚胺前体、低分子环状硅氧烷和溶剂的基础上,可以进一步包含追加成分。作为追加成分,可列举出例如表面活性剂和烷氧基硅烷化合物等。
[0460]
表面活性剂
[0461]
通过向本实施方式的树脂组合物中添加表面活性剂,从而能够提高树脂组合物的涂布性。具体而言,能够防止涂布膜中产生条纹。
[0462]
这种表面活性剂可列举出例如有机硅系表面活性剂、氟系表面活性剂、除此之外的非离子表面活性剂等。作为有机硅系表面活性剂,可列举出例如有机硅氧烷聚合物kf-640、642、643、kp341、x-70-092、x-70-093(商品名、信越化学工业公司制);sh-28pa、sh-190、sh-193、sz-6032、sf-8428、dc-57、dc-190(商品名、东丽道康宁有机硅公司制);silwet l-77、l-7001、fz-2105、fz-2120、fz-2154、fz-2164、fz-2166、l-7604(商品名、日本尤妮佳公司制);dbe-814、dbe-224、dbe-621、cms-626、cms-222、kf-352a、kf-354l、kf-355a、kf-6020、dbe-821、dbe-712(gelest)、byk-307、byk-310、byk-378、byk-333(商品名、byk-chemie japan公司制);granol(商品名、共荣社化学公司制)等。作为氟系表面活性剂,可列举出例如megafac f171、f173、r-08(大日本油墨化学工业公司制、商品名);fluorad fc4430、fc4432(住友3m公司、商品名)等。作为除此之外的非离子表面活性剂,可列举出例如聚氧乙烯月桂基醚、聚氧乙烯硬脂基醚、聚氧乙烯油烯基醚、聚氧乙烯辛基苯酚醚等。
[0463]
这些表面活性剂之中,从树脂组合物的涂布性(抑制条纹)的观点出发,优选为有机硅系表面活性剂、氟系表面活性剂,从降低固化工序时的氧浓度对yi值和总光线透射率造成的影响的观点出发,优选为有机硅系表面活性剂。在使用表面活性剂的情况下,其配混量相对于树脂组合物中的聚酰亚胺前体100质量份优选为0.001~5质量份、更优选为0.01~3质量份。
[0464]
烷氧基硅烷化合物
[0465]
将由本实施方式的树脂组合物得到的聚酰亚胺薄膜用于柔性基板等的情况下,从获得制造工艺中的支承体与聚酰亚胺薄膜的良好密合性的观点出发,树脂组合物可以含有相对于聚酰亚胺前体100质量份为0.01~20质量份的烷氧基硅烷化合物。通过使烷氧基硅烷化合物的含量相对于聚酰亚胺前体100质量份为0.01质量份以上,从而能够使支承体与聚酰亚胺薄膜之间得到良好的密合性。另外,从树脂组合物的保存稳定性的观点出发,烷氧基硅烷化合物的含量优选为20质量份以下。烷氧基硅烷化合物的含量相对于聚酰亚胺前体100质量份优选为0.02~15质量份、更优选为0.05~10质量份、进一步优选为0.1~8质量份。通过使用烷氧基硅烷化合物,从而在提高上述密合性的基础上,树脂组合物的涂布性提高(抑制条纹不均),并且,也能够降低固化时的氧浓度对聚酰亚胺薄膜的yi值造成的影响。
[0466]
作为烷氧基硅烷化合物,可列举出例如3-脲基丙基三乙氧基硅烷、双(2-羟基乙基)-3-氨基丙基三乙氧基硅烷、3-环氧丙氧基丙基三甲氧基硅烷、γ-氨基丙基三甲氧基硅烷、γ-氨基丙基三丙氧基硅烷、γ-氨基丙基三丁氧基硅烷、γ-氨基乙基三乙氧基硅烷、γ-氨基乙基三丙氧基硅烷、γ-氨基乙基三丁氧基硅烷、γ-氨基丁基三乙氧基硅烷、γ-氨基丁基三甲氧基硅烷、γ-氨基丁基三丙氧基硅烷、γ-氨基丁基三丁氧基硅烷、苯基硅烷三醇、三甲氧基苯基硅烷、三甲氧基(对甲苯基)硅烷、二苯基硅烷二醇、二甲氧基二苯基硅烷、
二乙氧基二苯基硅烷、二甲氧基二对甲苯基硅烷、三苯基硅烷醇、以及下述结构分别示出的烷氧基硅烷化合物等。
[0467][0468]
烷氧基硅烷化合物可以单独使用一种,也可以组合使用两种以上。
[0469]
《树脂组合物的制造方法》
[0470]
本实施方式中的树脂组合物的制造方法没有特别限定,可以利用例如以下的方法。
[0471]
〈含硅化合物的纯化〉
[0472]
本实施方式的树脂组合物可通过使包含酸二酐、二胺和含硅化合物的缩聚成分发生缩聚反应来制造。作为降低本实施方式的树脂组合物中包含的环状含硅化合物的总量的方法,可列举出:例如在缩聚反应之前,对含硅化合物进行纯化,降低环状含硅化合物的总量。或者,可以在缩聚反应之后,对树脂组合物进行纯化,降低环状含硅化合物的总量。
[0473]
作为对含硅化合物进行纯化的方法,可列举出:例如在任意容器内,边向含硅化合物中吹入非活性气体、例如氮气,边对其进行气提(stripping)。作为气提的温度,优选为200℃以上且300℃以下、更优选为220℃以上且300℃以下、进一步优选为240℃以上且300℃以下。作为气提的蒸气压,越低越优选,为1000pa以下、更优选为300pa以下、进一步优选为200pa以下、更进一步优选为133.32pa(1mmhg)以下。作为气提的时间,优选为4小时以上且12小时以下、更优选为6小时以上且10小时以下。通过调整至上述条件而能够有效地去除单环状的含硅化合物,另外,能够将环状含硅化合物的总量控制至优选范围。
[0474]
〈聚酰亚胺/聚酰亚胺前体的合成〉
[0475]
本实施方式的聚酰亚胺前体可通过使包含酸二酐、二胺和含硅化合物的缩聚成分发生缩聚反应来合成。
[0476]
关于第一方式所述的聚酰亚胺/聚酰亚胺前体的合成,提供例如包括如下的任意工序的树脂组合物的制造方法。
[0477]
·
使上述说明的通式(8)所示的化合物或通式(9)所示的化合物与通式(10)所示的含硅化合物发生缩聚反应而得到聚酰亚胺后,使其与其它化合物发生缩聚反应,从而提
供包含聚酰亚胺前体和聚酰亚胺的树脂组合物的工序;
[0478]
·
使上述说明的二胺或酸二酐与通式(10)所示的含硅化合物发生缩聚反应而得到聚酰亚胺后,使其与其它化合物发生缩聚反应,从而提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物的工序。
[0479]
关于第二方式所述的聚酰亚胺/聚酰亚胺前体的合成,提供例如包括如下的任意工序的树脂组合物的制造方法。
[0480]
·
使上述说明的通式(3)所示的化合物与通式(4)所示的化合物与通式(5)所示的含硅化合物与其它化合物发生缩聚反应,提供包含聚酰亚胺前体或聚酰亚胺的树脂组合物的工序;
[0481]
·
使上述说明的通式(3)所示的化合物与其它化合物与通式(4)所示的化合物与其它化合物发生缩聚反应而得到聚酰亚胺后,使上述说明的通式(5)所示的含硅化合物与其它化合物发生缩聚反应,从而提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物的工序;或者
[0482]
·
使上述说明的二胺与酸二酐发生缩聚反应而得到聚酰亚胺后,使通式(5)所示的含硅化合物与其它化合物发生缩聚反应,提供包含聚酰亚胺前体和聚酰亚胺的树脂组合物的工序。
[0483]
另外,含硅化合物优选使用进行上述纯化而得到的物质。在优选方式中,缩聚成分由酸二酐、二胺和含硅化合物形成。缩聚反应优选在适当的溶剂中进行。具体而言,可列举出:例如,在使规定量的二胺成分和含硅化合物溶解于溶剂后,向所得二胺溶液中添加规定量的酸二酐,并进行搅拌的方法。
[0484]
关于合成聚酰亚胺/聚酰亚胺前体时的酸二酐与二胺的摩尔比,从聚酰亚胺前体树脂的高分子量化、树脂组合物的狭缝涂布特性的观点出发,优选为酸二酐:二胺=100:90~100:110(相对于酸二酐1摩尔份,二胺为0.90~1.10摩尔份)的范围,更优选为100:95~100:105(相对于酸二酐1摩尔份,二胺为0.95~1.05摩尔份)的范围。
[0485]
聚酰亚胺/聚酰亚胺前体的分子量可通过调整酸二酐、二胺和含硅化合物的种类、酸二酐与二胺的摩尔比、添加封端剂、调整反应条件等来控制。酸二酐成分与二胺成分的摩尔比越接近1:1,且封端剂的用量越少,则越能够使聚酰亚胺前体发生高分子量化。
[0486]
作为酸二酐成分和二胺成分,推荐使用高纯度品。作为其纯度,分别优选为98质量%以上、更优选为99质量%以上、进一步优选为99.5质量%以上。通过降低酸二酐成分和二胺成分中的水分含量,从而也能够高纯度化。在使用多种酸二酐成分和/或多种二胺成分的情况下,优选以酸二酐成分整体计和以二胺成分整体计具有上述纯度,更优选所使用的全部种类的酸二酐成分和二胺成分分别具有上述纯度。
[0487]
作为反应的溶剂,只要是能够溶解酸二酐成分和二胺成分、以及生成的聚酰亚胺/聚酰亚胺前体,且能够得到高分子量聚合物的溶剂,就没有特别限定。作为这种溶剂,可列举出例如非质子性溶剂、酚系溶剂、醚和二醇系溶剂等。
[0488]
作为非质子性溶剂,可列举出例如n,n-二甲基甲酰胺(dmf)、n,n-二甲基乙酰胺(dmac)、n-甲基-2-吡咯烷酮(nmp)、n-甲基己内酰胺、1,3-二甲基咪唑啉酮、四甲基脲、以及下述通式的酰胺系溶剂;γ-丁内酯、γ-戊内酯等内酯系溶剂;六甲基磷酰胺、六甲基膦三酰胺等含磷系酰胺系溶剂;二甲基砜、二甲基亚砜、环丁砜等含硫系溶剂;环己酮、甲基环己
酮等酮系溶剂;甲基吡啶、吡啶等叔胺系溶剂;乙酸(2-甲氧基-1-甲基乙基)酯等酯系溶剂等。
[0489][0490]
{式中,r
12
=甲基所示的
エクアミド
m100(商品名:出光兴产公司制)和r
12
=正丁基所示的
エクアミド
b100(商品名:出光兴产公司制)}
[0491]
作为酚系溶剂,可列举出例如苯酚、邻甲酚、间甲酚、对甲酚、2,3-二甲苯酚、2,4-二甲苯酚、2,5-二甲苯酚、2,6-二甲苯酚、3,4-二甲苯酚、3,5-二甲苯酚等。
[0492]
作为醚和二醇系溶剂,可列举出例如1,2-二甲氧基乙烷、双(2-甲氧基乙基)醚、1,2-双(2-甲氧基乙氧基)乙烷、双[2-(2-甲氧基乙氧基)乙基]醚、四氢呋喃、1,4-二噁烷等。
[0493]
这些溶剂可以单独使用或混合使用两种以上。
[0494]
在聚酰亚胺/聚酰亚胺前体的合成中使用的溶剂在常压下的沸点优选为60~300℃、更优选为140~280℃、进一步优选为170~270℃。通过使溶剂的沸点小于300℃,从而干燥工序变为短时间。若溶剂的沸点为60℃以上,则在干燥工序中不易发生树脂膜表面的粗糙的发生、气泡向树脂膜中的混入等,能够得到更均匀的薄膜。尤其是,从溶解性和降低涂布时的边缘异常的观点出发,优选使用沸点为170~270℃和/或20℃下的蒸气压为250pa以下的溶剂。更具体而言,优选为选自由n-甲基-2-吡咯烷酮(nmp)、γ-丁内酯(gbl)和第一方式和第二方式所述的通式(6)所示的化合物组成的组中的1种以上。
[0495]
为了良好地进行缩聚反应,溶剂中的水分含量例如优选为3,000质量ppm以下。本实施方式的树脂组合物中,分子量小于1,000的分子的含量优选小于5质量%。认为树脂组合物中存在分子量小于1,000的分子是因为:与在合成时使用的溶剂、原料(酸二酐、二胺)的水分量有关。即,可认为这是因为:一部分酸二酐单体的酸酐基因水分而发生水解,形成羧基,不会高分子量化,而是以低分子的状态残留。因此,上述缩聚反应中使用的溶剂的水分量越少越优选。溶剂的水分量优选设为3,000质量ppm以下,更优选设为1,000质量ppm以下。同样地,关于原料中包含的水分量,也优选设为3,000质量ppm以下,更优选设为1,000质量ppm以下。
[0496]
可以认为:溶剂的水分量与所使用的溶剂级别(脱水级别、通用级别等)、溶剂容器(瓶、18l罐、小罐等)、溶剂的保管状态(是否封入稀有气体等)、从开封起至使用为止的时间(是否在开封后立即使用、或者是否在开封后经过一定时间后再使用等)等有关。可以认为:与合成前的反应器的稀有气体置换、在合成中是否流通稀有气体等也有关。因此推荐的是:在合成聚酰亚胺前体时,使用高纯度品作为原料,使用水分量少的溶剂,并且,采取在反应前和反应中不使来自环境的水分向体系内混入那样的措施。
[0497]
在溶剂中溶解各缩聚成分时,根据需要可以进行加热。从得到聚合度高的聚酰亚胺前体的观点出发,作为合成聚酰亚胺前体时的反应温度,可优选为0℃~120℃、40℃~100℃或60~100℃,作为聚合时间,可优选为1~100小时或2~10小时。通过将聚合时间设为1小时以上,从而形成聚合度均匀的聚酰亚胺前体,通过设为100小时以下,能够得到聚合度高的聚酰亚胺前体。
[0498]
本实施方式的树脂组合物中,除了包含本实施方式中的聚酰亚胺/聚酰亚胺前体
之外,可以包含其它追加的聚酰亚胺前体。然而,从降低聚酰亚胺薄膜的yi值和总光线透射率的氧依赖性的观点出发,追加的聚酰亚胺/聚酰亚胺前体的质量比例相对于树脂组合物中的聚酰亚胺/聚酰亚胺前体的总量优选为30质量%以下、进一步优选为10质量%以下。
[0499]
本实施方式中的聚酰亚胺前体的一部分可以进行了酰亚胺化(部分酰亚胺化)。通过对聚酰亚胺前体进行部分酰亚胺化,从而能够提高保存树脂组合物时的粘度稳定性。从树脂组合物中的聚酰亚胺前体的溶解性与溶液的保存稳定性呈现平衡的观点出发,此时的酰亚胺化率优选为5%以上、更优选为8%以上,且优选为80%以下、更优选为70%以下、进一步优选为50%以下。该部分酰亚胺化通过将聚酰亚胺前体加热并进行脱水闭环而得到。该加热可以在优选120~200℃、更优选150~185℃、进一步优选150~180℃的温度下进行优选15分钟~20小时、更优选30分钟~10小时。
[0500]
作为本实施方式的聚酰亚胺前体,可以使用通过向利用上述反应而得到的聚酰亚胺/聚酰亚胺前体中添加n,n-二甲基甲酰胺二甲基缩醛或n,n-二甲基甲酰胺二乙基缩醛并加热而使一部分或全部羧酸发生酯化而得到的物质。通过酯化而能够提高保存时的粘度稳定性。这些酯改性聚酰胺酸也可以通过使上述酸二酐成分依次与相对于酸酐基为1当量的一元醇、以及亚硫酰氯、二环己基碳二亚胺等脱水缩合剂发生反应后,再与二胺成分发生缩合反应的方法来获得。
[0501]
〈聚酰亚胺的合成〉
[0502]
作为更优选的方式,聚酰亚胺清漆可通过将酸二酐成分和二胺成分溶解于溶剂、例如有机溶剂,添加甲苯等共沸溶剂,将在酰亚胺化时产生的水排除至体系外,从而以含有聚酰亚胺和溶剂的聚酰亚胺溶液(也称为聚酰亚胺清漆)的形式来制造。此处,反应时的条件没有特别限定,例如,反应温度为0℃~180℃、反应时间为3~72小时。为了与含有磺基的二胺类充分进行反应,优选以180℃进行12小时左右的加热反应。另外,在反应时,优选为氩气、氮气等非活性气氛。
[0503]
〈树脂组合物的制备〉
[0504]
在合成聚酰亚胺前体时使用的溶剂与树脂组合物中含有的溶剂相同的情况下,可以将所合成的聚酰亚胺/聚酰亚胺前体溶液直接用作树脂组合物。根据需要,通过在室温(25℃)~80℃的温度范围内向聚酰亚胺前体中添加进一步的溶剂和追加成分中的1种以上,并进行搅拌混合,从而也可以制备树脂组合物。该搅拌混合可使用具备搅拌叶片的three one motor(新东化学公司制)、自转公转搅拌器等适当装置来进行。根据需要,可以将树脂组合物加热至40℃~100℃。
[0505]
另一方面,在合成聚酰亚胺/聚酰亚胺前体时使用的溶剂与树脂组合物中含有的溶剂不同的情况下,可通过例如再沉淀、溶剂馏去等适当方法将所合成的聚酰亚胺前体溶液中的溶剂去除而分离出聚酰亚胺/聚酰亚胺前体。接着,通过在室温(25℃)~80℃的温度范围内向分离出的聚酰亚胺前体中添加期望的溶剂和根据需要而追加的成分,并进行搅拌混合,从而也可以制备树脂组合物。
[0506]
在如上所述地制备树脂组合物后,通过将树脂组合物以例如130~200℃加热例如5分钟~2小时,从而可以以聚合物不发生析出的程度对聚酰亚胺前体的一部分进行脱水酰亚胺化(部分酰亚胺化)。通过控制加热温度和加热时间,从而能够控制酰亚胺化率。通过对聚酰亚胺前体进行部分酰亚胺化,从而能够提高对树脂组合物加以保存时的粘度稳定性。
[0507]
从狭缝涂布性能的观点出发,树脂组合物的溶液粘度优选为500~100,000mpa
·
s、更优选为1,000~50,000mpa
·
s、进一步优选为3,000~20,000mpa
·
s。具体而言,从不易自狭缝喷嘴中漏液的观点出发,优选为500mpa
·
s以上、更优选为1,000mpa
·
s以上、进一步优选为3,000mpa
·
s以上。从狭缝喷嘴难以堵塞的观点出发,优选为100,000mpa
·
s以下、更优选为50,000mpa
·
s以下、进一步优选为20,000mpa
·
s以下。
[0508]
关于合成聚酰亚胺/聚酰亚胺前体时的树脂组合物的溶液粘度,若高于200,000mpa
·
s,则有可能产生在合成时难以搅拌的问题。其中,即便在合成时溶液达到高粘度,通过在反应结束后添加溶剂并进行搅拌,从而也能够得到处理性良好的粘度的树脂组合物。本实施方式中的树脂组合物的溶液粘度是使用e型粘度计(例如visconicehd、东机产业公司制)并在23℃下测得的值。
[0509]
从保存树脂组合物时的粘度稳定性的观点出发,本实施方式的树脂组合物的水分量优选为3,000质量ppm以下、更优选为2,500质量ppm以下、进一步优选为2,000质量ppm以下、更进一步优选为1,500质量ppm以下、特别优选为1,000质量ppm以下、特别优选为500质量ppm以下、特别优选为300质量ppm以下、特别优选为100质量ppm以下。
[0510]
《聚酰亚胺薄膜及其制造方法》
[0511]
使用本实施方式的树脂组合物,能够提供聚酰亚胺薄膜(以下也称为聚酰亚胺树脂膜)。本实施方式的聚酰亚胺薄膜的制造方法包括:在支承体的表面上涂布本实施方式的树脂组合物的涂布工序;将上述树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及将上述聚酰亚胺树脂膜从上述支承体上剥离的剥离工序。
[0512]
〈涂布工序〉
[0513]
在涂布工序中,在支承体的表面上涂布本实施方式的树脂组合物。支承体只要对于其后的成膜工序(加热工序)中的加热温度具有耐热性,且剥离工序中的剥离性良好,就没有特别限定。作为支承体,可列举出例如玻璃基板、例如无碱玻璃基板;硅晶圆;pet(聚对苯二甲酸乙二醇酯)、opp(拉伸聚丙烯)、聚乙二醇对苯二甲酸酯、聚萘二甲酸乙二醇酯、聚碳酸酯、聚酰亚胺、聚酰胺酰亚胺、聚醚酰亚胺、聚醚醚酮、聚醚砜、聚苯砜、聚苯硫醚等树脂基板;不锈钢、氧化铝、铜、镍等金属基板等。
[0514]
在形成薄膜状的聚酰亚胺成形体的情况下,优选为例如玻璃基板、硅晶圆等,在形成厚膜状的膜状或片状的聚酰亚胺成形体的情况下,优选为例如由pet(聚对苯二甲酸乙二醇酯)、opp(拉伸聚丙烯)等形成的支承体。
[0515]
作为涂布方法,通常可列举出刮板刀涂机、气刀涂布机、辊涂机、旋转涂布机、流涂机、模涂机、棒涂机等涂布方法;旋涂、喷涂、浸涂等涂布方法;丝网印刷和凹版印刷等所代表的印刷技术等。本实施方式的树脂组合物优选基于狭缝涂布的涂布。涂布厚度应该根据树脂薄膜的期望厚度和树脂组合物中的聚酰亚胺前体的含量来适当调整,优选为1~1,000μm左右。涂布工序中的温度可以为室温,为了降低粘度而改善作业性,可以将树脂组合物加热至例如40~80℃。
[0516]
〈任选的干燥工序〉
[0517]
可以在涂布工序后接着进行干燥工序,或者,可以省略干燥工序而直接推进至后续的成膜工序(加热工序)。干燥工序是出于去除树脂组合物中的有机溶剂的目的而进行的。在进行干燥工序的情况下,可以使用例如加热板、箱型干燥机、传送带型干燥机等适当
装置。干燥工序的温度优选为80~200℃、更优选为100~150℃。干燥工序的实施时间优选为1分钟~10小时、更优选为3分钟~1小时。如上那样操作,在支承体上形成含有聚酰亚胺前体的涂膜。
[0518]
〈成膜工序〉
[0519]
接着,进行成膜工序(加热工序)。加热工序是进行上述涂膜中包含的有机溶剂的去除,且进行涂膜中的聚酰亚胺前体的酰亚胺化反应而得到聚酰亚胺树脂膜的工序。该加热工序可使用例如惰性气体烘箱、加热板、箱型干燥机、传动带型干燥机等装置来进行。该工序可以与干燥工序同时进行,也可以依次进行两个工序。
[0520]
加热工序可以在空气气氛下进行,从得到安全性、以及所得聚酰亚胺薄膜的良好透明性、低厚度方向延迟(rth)和低yi值的观点出发,优选在非活性气体气氛下进行。作为非活性气体,可列举出例如氮气、氩气等。加热温度可根据聚酰亚胺前体的种类和树脂组合物中的溶剂种类来适当设定,优选为250℃~550℃、更优选为300~450℃。如果为250℃以上,则良好地进行酰亚胺化,如果为550℃以下,则能够避免所得聚酰亚胺薄膜的透明性降低、耐热性恶化等不良情况。加热时间优选为0.1~10小时左右。
[0521]
本实施方式中,从所得聚酰亚胺薄膜的透明性和yi值的观点出发,上述加热工序中的周围气氛的氧浓度优选为2,000质量ppm以下、更优选为100质量ppm以下、进一步优选为10质量ppm以下。通过在氧浓度为2,000质量ppm以下的气氛中进行加热,从而能够将所得聚酰亚胺薄膜的yi值设为30以下。
[0522]
〈剥离工序〉
[0523]
在剥离工序中,将支承体上的聚酰亚胺树脂膜冷却至例如室温(25℃)~50℃左右后再进行剥离。作为该剥离工序,可列举出例如下述(1)~(4)的方式。
[0524]
(1)在通过上述方法而制作包含聚酰亚胺树脂膜/支承体的构成体后,从结构体的支承体侧照射激光,对支承体与聚酰亚胺树脂膜的界面进行磨蚀加工,由此剥离聚酰亚胺树脂的方法。作为激光的种类,可列举出固体(yag)激光、气体(uv准分子)激光等。优选使用波长为308nm等的光谱(参照日本特表2007-512568号公报、日本特表2012-511173号公报等)。
[0525]
(2)在支承体上涂布树脂组合物之前,在支承体上形成剥离层,其后得到包含聚酰亚胺树脂膜/剥离层/支承体的构成体,并剥离聚酰亚胺树脂膜的方法。作为剥离层,可列举出parylene(注册商标、日本parylene合同会社制)、氧化钨,可以使用植物油系、有机硅系、氟系、醇酸系等的脱模剂(参照日本特开2010-067957号公报、日本特开2013-179306号公报等)。
[0526]
也可以组合使用该方法(2)和方法(1)的激光照射。
[0527]
(3)使用能够蚀刻的金属基板作为支承体,在得到包含聚酰亚胺树脂膜/支承体的构成体后,利用蚀刻剂对金属进行蚀刻,由此得到聚酰亚胺树脂薄膜的方法。作为金属,可以使用例如铜(作为具体例,为三井金属矿业公司制的电解铜箔“dff”)、铝等。作为蚀刻剂,可以对铜使用氯化铁等,对铝使用稀盐酸等。
[0528]
(4)通过上述方法而得到包含聚酰亚胺树脂膜/支承体的构成体后,在聚酰亚胺树脂膜表面粘贴粘合薄膜,从支承体中分离出粘合薄膜/聚酰亚胺树脂膜,其后,从粘合薄膜中分离出聚酰亚胺树脂膜的方法。
[0529]
这些剥离方法之中,从所得聚酰亚胺树脂薄膜的表背的折射率之差、yi值和伸长率的观点出发,优选为方法(1)或(2)。从所得聚酰亚胺树脂薄膜的表背的折射率之差的观点出发,更优选方法(1)、即在剥离工序之前进行从支承体侧照射激光的照射工序。需要说明的是,在方法(3)中使用铜作为支承体时,观察到所得聚酰亚胺树脂薄膜的yi值变大、伸长率变小的倾向。可认为这是铜离子的影响。
[0530]
所得聚酰亚胺薄膜的厚度没有限定,优选为1~200μm、更优选为5~100μm。
[0531]
《聚酰亚胺薄膜的用途》
[0532]
由本实施方式的树脂组合物得到的聚酰亚胺薄膜可用作例如半导体绝缘膜、薄膜晶体管液晶显示器(tft-lcd)绝缘膜、电极保护膜,另外,可用作液晶显示器、有机电致发光显示器、场致发射显示器、电子纸等显示装置的透明基板等。尤其是,由本实施方式的树脂组合物得到的聚酰亚胺薄膜可以在柔性设备的制造中适宜地用作薄膜晶体管(tft)基板、滤色器基板、触摸面板基板、透明导电膜(ito、indium thin oxide)的基板。作为可应用本实施方式中的聚酰亚胺薄膜的柔性设备,可列举出例如柔性显示器用tft设备、柔性太阳能电池、柔性触摸面板、柔性照明、柔性电池、柔性印刷基板、柔性滤色器、智能手机用途的表面覆盖镜头等。
[0533]
典型而言,在使用聚酰亚胺薄膜得到的柔性基板上形成tft的工序在150~650℃的大范围的温度下实施。具体而言,在制作使用非晶硅得到的tft设备的情况下,通常需要250℃~350℃的工艺温度,本实施方式的聚酰亚胺薄膜需要可耐受该温度的必要,因此,具体而言,需要适当选择具有工艺温度以上的玻璃化转变温度、热分解开始温度的聚合物结构。
[0534]
在制作使用金属氧化物半导体(igzo等)得到的tft设备的情况下,通常需要320℃~400℃的工艺温度,本实施方式的聚酰亚胺薄膜需要能够耐受该温度,因此,需要适当选择具有tft制作工艺最高温度以上的玻璃化转变温度、热分解开始温度的聚合物结构。
[0535]
在制作使用低温多晶硅(ltps)得到的tft设备的情况下,通常需要380℃~520℃的工艺温度,本实施方式的聚酰亚胺薄膜需要能够耐受该温度,因此,需要适当选择tft制作工艺最高温度以上的玻璃化转变温度、热分解开始温度。另一方面,由于这些热历程,从而存在越是暴露于高温工序则聚酰亚胺薄膜的光学特性(尤其是光线透射率、延迟特性和yi值)越会降低的倾向。但是,由本实施方式的聚酰亚胺前体得到的聚酰亚胺即便历经热历程也具有良好的光学特性。
[0536]
以下,作为本实施方式的聚酰亚胺薄膜的用途例,针对显示器和层叠体的制造方法进行说明。
[0537]
〈显示器的制造方法〉
[0538]
本实施方式的显示器的制造方法包括:在支承体的表面上涂布本实施方式的树脂组合物的涂布工序;将上述树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;在上述聚酰亚胺树脂膜上形成元件的元件形成工序;以及将形成有上述元件的上述聚酰亚胺树脂膜从上述支承体上剥离的剥离工序。
[0539]
柔性有机el显示器的制造例
[0540]
图1是示出作为本实施方式的显示器的例子的顶部发光型柔性有机el显示器的比聚酰亚胺基板更靠上部的结构的示意图。针对图1的有机el结构部25进行说明。例如,将发
出红色光的有机el元件250a、发出绿色光的有机el元件250b和发出蓝色光的有机el元件250c作为1个单元,并排列成矩阵状,利用分隔壁(堤)251来划分各有机el元件的发光区域。各有机el元件由下部电极(阳极)252、空穴传输层253、发光层254、上部电极(阴极)255构成。在表示由氮化硅(sin)、氧化硅(sio)形成的cvd多层膜(多层阻挡层)的下部层2a上,设置有多个用于驱动有机el元件的tft256(选自低温多晶硅(ltps)、金属氧化物半导体(igzo等))、具备接触孔257的层间绝缘膜258、以及下部电极259。有机el元件被密封基板2b密封,在各有机el元件与密封基板2b之间形成有中空部261。
[0541]
柔性有机el显示器的制造工序包括:在玻璃基板支承体上制作聚酰亚胺薄膜,在其上部制造图1所示的有机el基板的工序;制造密封基板的工序;将两个基板贴合的组装工序;以及,从玻璃基板支承体上剥离在聚酰亚胺薄膜上制作的有机el显示器的剥离工序。有机el基板制造工序、密封基板制造工序和组装工序可以应用公知的制造工序。以下列举出其中的一例,但不限定于此。剥离工序与上述聚酰亚胺薄膜的剥离工序相同。
[0542]
例如,如果参照图1,则首先通过上述方法在玻璃基板支承体上制作聚酰亚胺薄膜,通过cvd法、溅射法,在其上部制作由氮化硅(sin)和氧化硅(sio)的多层结构形成的多层阻挡层(图1中的下部基板2a),使用光致抗蚀剂等,在其上部制作用于驱动tft的金属布线层。使用cvd法,在其上部制作sio等的有源缓冲层,在其上部制作金属氧化物半导体(igzo)、低温多晶硅(ltps)等tft设备(图1中的tft256)。在制作柔性显示器用tft基板后,利用感光性丙烯酸类树脂等来形成具备接触孔257的层间绝缘膜258。利用溅射法等而成膜出ito膜,以与tft配对的方式形成下部电极259。
[0543]
接着,利用感光性聚酰亚胺等来形成分隔壁(堤)251后,在被分隔壁划分出的各空间内形成空穴传输层253、发光层254。以覆盖发光层254和分隔壁(堤)251的方式形成上部电极(阴极)255。其后,以精细金属掩模等作为掩模,利用公知方法来蒸镀发出红色光的有机el材料(与图1中的发出红色光的有机el元件250a对应)、发出绿色光的有机el材料(与图1中的发出绿色光的有机el元件250b对应)和发出蓝色光的有机el材料(与图1中的发出蓝色光的有机el元件250c对应),由此制作有机el基板。通过利用密封薄膜等(图1中的密封基板2b)来密封有机el基板,并利用激光剥离等公知的剥离方法从玻璃基板支承体上剥离比聚酰亚胺基板更靠上部的设备,由此能够制作顶部发光形柔性有机el显示器。在使用本实施方式的聚酰亚胺的情况下,可以制作透明(see through)型的柔性有机el显示器。也可以利用公知方法来制作底部发光形的柔性有机el显示器。
[0544]
柔性液晶显示器的制造例
[0545]
可使用本实施方式的聚酰亚胺薄膜来制作柔性液晶显示器。作为具体的制作方法,利用上述方法在玻璃基板支承体上制作聚酰亚胺薄膜,使用上述方法来制作由例如非晶硅、金属氧化物半导体(igzo等)和低温多晶硅形成的tft基板。另行按照本实施方式的涂布工序和成膜工序,在玻璃基板支承体上制作聚酰亚胺薄膜,按照公知方法并使用彩色光阻等,制作具备聚酰亚胺薄膜的滤色器玻璃基板(cf基板)。在tft基板和cf基板中的一者上,通过丝网印刷将包含热固性环氧树脂等的密封材料涂布成液晶注入口的部分缺损的框状图案,使另一个基板保持与液晶层的厚度相当的直径,散布由塑料或二氧化硅形成的球状间隔物。
[0546]
接着,将tft基板与cf基板进行贴合,使密封材料发生固化。并且,通过减压法,向
被tft基板和cf基板以及密封材料包围的空间内注入液晶材料,对液晶注入口涂布热固化树脂,通过加热而将液晶材料密封,由此形成液晶层。最后,通过激光剥离法等将cf侧的玻璃基板和tft侧的玻璃基板在聚酰亚胺薄膜与玻璃基板的界面处进行剥离,由此能够制作柔性液晶显示器。
[0547]
〈层叠体的制造方法〉
[0548]
本实施方式的层叠体的制造方法包括:在支承体的表面上涂布本实施方式的树脂组合物的涂布工序;将上述树脂组合物加热而形成聚酰亚胺树脂膜的成膜工序;以及,在上述聚酰亚胺树脂膜上形成元件的元件形成工序。
[0549]
作为层叠体中的元件,可列举出在上述柔性设备的制造中例示出的元件。作为支承体,可以使用例如玻璃基板。涂布工序和成膜工序的优选的具体步骤与针对上述聚酰亚胺薄膜的制造方法而记载的步骤相同。在元件形成工序中,在形成在支承体上的作为柔性基板的聚酰亚胺树脂膜上形成上述元件。其后,可任选在剥离工序中将聚酰亚胺树脂膜和元件从支承体上剥离。
[0550]
实施例
[0551]
以下,通过实施例和比较例来具体说明本发明的实施方式,但本发明不限定于这些实施例和比较例。
[0552]
〔第一方式〕
[0553]
《测定和评价方法》
[0554]
〈重均分子量〉
[0555]
重均分子量(mw)和数均分子量(mn)利用凝胶渗透色谱法(gpc),并利用下述条件来测定。
[0556]
作为溶剂,使用nmp(和光纯药工业公司制、高效液相色谱用、在即将测定之前添加24.8mmol/l的溴化锂一水合物(和光纯药工业公司制、纯度为99.5%)和63.2mmol/l的磷酸(和光纯药工业公司制、高效液相色谱用)并溶解而得到的物质)。用于计算重均分子量的标准曲线使用标准聚苯乙烯(东曹公司制)来制作。
[0557]
柱:shodex kd-806m(昭和电工公司制)
[0558]
流速:1.0ml/分钟
[0559]
柱温度:40℃
[0560]
泵:pu-2080plus(jasco公司制)
[0561]
检测器:ri-2031plus(ri:差示折射计、jasco公司制)和uv-2075plus(uv-vis:紫外可见吸光度计、jasco公司制)
[0562]
《组合物(清漆)的粘度稳定性的评价》
[0563]
将实施例和比较例中制备的树脂组合物在23℃、50%rh的气氛下放置24小时,使用带有调温机的粘度计(东机产业公司制的viscomater tve-35h),测定树脂组合物的初始(0天)的粘度。并且,将树脂组合物在23℃、50%rh的气氛下放置10天,与初始同样操作,测定10天后的粘度。接着,计算下述式所示的粘度稳定性。
[0564]
粘度稳定性(%/day)=|(初始粘度(mpa
·
s))-(10天后的粘度(mpa
·
s))|/(初始粘度(mpa
·
s))
×
100/10(day)
[0565]
粘度稳定性按照下述基准进行评价。
[0588]
《全部树脂中的含硅化合物的比例》
[0589]
如下所述地计算实施例/比较例的树脂组合物的全部树脂的总质量中的含硅化合物的比例(质量%)。
[0590]
含硅化合物的比例(质量%)=含硅化合物的质量(g)/各单体(酸二酐单体、二胺单体、含硅化合物)的质量的总量(g)
×
100
[0591]
另外,含硅化合物的比例可使用清漆并利用下述方法来求出。
[0592]
向清漆中添加适当量的水,以80℃进行3天的加热处理,使酸成分、胺成分和解聚成分发生解聚,制成酸单体和胺单体。其后,通过馏去溶剂而得到混合有酸单体和胺单体的粉体,制备乙腈溶液,进行高效液相色谱质谱分析(lc/ms)测定。并且,求出各单体的峰面积,可以根据其峰面积比进行计算。
[0593]
《酰亚胺化率》
[0594]
如下所述地计算实施例/比较例的树脂的酰亚胺化率(%)。
[0595]
(通式(10)的l1和l2为氨基的情况)
[0596]
酰亚胺化率(%)=
[0597]
酰亚胺化工序的二胺单体(包括通式(10)的l1和l2为氨基的含硅化合物)的总摩尔数/{酰亚胺化工序的二胺单体(包括通式(10)的l1和l2为氨基的含硅化合物)的总摩尔数+酰胺化工序的二胺单体(包括通式(10)的l1和l2为氨基的含硅化合物)的总摩尔数}
×
100
[0598]
例如,在实施例i-1的情况下,
[0599]
酰亚胺化工序的二胺单体(包括通式(10)的l1和l2为氨基的含硅化合物)的总摩尔数:93.6mmol
[0600]
酰胺化工序的二胺单体(包括通式(10)的l1和l2为氨基的含硅化合物)的总摩尔数:111.9mmol
[0601]
酰亚胺化率成为83.7%。
[0602]
(通式(10)的l1和l2为酸酐基的情况)
[0603]
酰亚胺化率(%)=
[0604]
酰亚胺化工序的酸二酐单体(包括通式(10)的l1和l2为酸酐基的含硅化合物)的总摩尔数/{酰亚胺化工序的酸酐单体(包括通式(10)的l1和l2为酸酐基的含硅化合物)的总摩尔数+酰胺化工序的酸二酐单体(包括通式(10)的l1和l2为酸酐基的含硅化合物)的总摩尔数}
×
100
[0605]
另外,酰亚胺化率可使用清漆并利用下述方法来求出。
[0606]
向清漆中添加适当量的水,以80℃进行3天的加热处理,使酸成分、胺成分和解聚成分发生解聚,制成酸单体和胺单体。其后,通过馏去溶剂而得到混合有酸单体和胺单体的粉体,制备乙腈溶液,进行高效液相色谱质谱分析(lc/ms)测定。并且,求出各单体的峰面积,可以根据其峰面积比进行计算。
[0607]
另外,酰亚胺化率也可以使用ir(红外分光光度计)进行测定。在将清漆利用水溶剂进行再沉淀后,对粉体进行分离干燥,然后添加kbr而制成粒料,用作样品。并且,通过利用一次反射atr法来测定树脂层的红外线吸收光谱,从而可以以1009cm-1
的苯环碳氢键作为基准,根据1778cm-1
的源自酰亚胺基的吸光度进行计算。此处,将在400℃下对清漆进行1小时热处理后的聚酰亚胺薄膜的酰亚胺化率设为100%。
[0608]
《二胺中的含硅化合物比例酰亚胺单元与酰胺单元之差》
[0609]
实施例/比较例的树脂的二胺中的含硅化合物比例的、酰亚胺单元与酰胺单元之差根据下述式来求出。
[0610]
a:酰亚胺单元的二胺中的含硅化合物比例(质量%)=在酰亚胺化工序中使用的含硅化合物/在酰亚胺化工序中使用的二胺单体(包括含硅化合物)的质量的总量
×
100
[0611]
b:酰胺单元的二胺中的含硅化合物比例(质量%)=在酰亚胺化工序中使用的含硅化合物/在酰胺化工序中使用的二胺单体(包括含硅化合物)的质量的总量
×
100
[0612]
a也可以换称为“构成通式(7)中的p5的二胺之中的通式(10)的比例(质量%)”。
[0613]
另外,b也可以换称为“构成通式(6)中的p3的二胺之中的通式(10)的比例(质量%)”。
[0614]
并且,关于二胺中的含硅化合物比例,酰亚胺单元与酰胺单元之差示作“b-a”。
[0615]
将实施例i-14、i-16、i-17、比较例i-4~i-6的b-a的值、以及它们的清漆和聚酰亚胺薄膜的特性示于表5。
[0616]
《实施例i-1》
[0617]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(189g)、甲苯(19g)、作为二胺的daba(73.0mmol、11.1g)、含硅化合物(2)(6.699mmol、10.72g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(18.3mmol、2.8g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液(以下也称为清漆)。
[0618]
《实施例i-2》
[0619]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(191g)、甲苯(19g)、作为二胺的daba(13.6g)、含硅化合物(1)(10.82g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(0.7g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0620]
《实施例i-3》
[0621]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(191g)、甲苯(19g)、作为二胺的daba(7.3g)、含硅化合物(3)(10.85g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(7.3g)。酸二酐与二胺的摩尔比为100:98。并
且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0622]
《实施例i-4》
[0623]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(191g)、甲苯(19g)、作为二胺的daba(8.7g)、含硅化合物(4)(10.85g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(5.8g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0624]
《实施例i-5》
[0625]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(186g)、甲苯(19g)、作为二胺的daba(14.9g),接着,在室温下添加作为酸二酐的cpoda(32.8g)、含硅化合物(5)(10.51g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为酸二酐的cpoda(3.6g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0626]
《实施例i-6》
[0627]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(184g)、甲苯(18g)、作为二胺的daba(12.9g)、含硅化合物(1)(10.44g),接着,在室温下添加作为酸二酐的cpoda(28.8g)、odpa(7.8g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(1.4g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0628]
《实施例i-7》
[0629]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(194g)、甲苯(19g)、作为二胺的daba(11.1g)、含硅化合物(2)(11.02g),接着,在室温下添加作为酸二酐的cpoda(28.8g)、6fda(11.1g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(2.8g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的
nmp溶液。
[0630]
《实施例i-8》
[0631]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(197g)、甲苯(20g)、作为二胺的daba(8.6g)、含硅化合物(1)(11.19g),接着,在室温下添加作为酸二酐的cpoda(28.8g)、bpaf(11.5g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(5.7g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0632]
《实施例i-9》
[0633]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(193g)、甲苯(19g)、作为二胺的daba(7.2g)、含硅化合物(1)(10.92g),接着,在室温下添加作为酸二酐的cpoda(28.8g)、bzda(10.1g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(7.2g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0634]
《实施例i-10》
[0635]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(170g)、甲苯(17g)、作为二胺的daba(13.3g)、含硅化合物(2)(9.62g),接着,在室温下添加作为酸二酐的cpoda(28.8g)、bnbdf(4.1g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(0.7g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0636]
《实施例i-11》
[0637]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(204g)、甲苯(20g)、作为二胺的daba(10.3g)、bafl(5.9g)、含硅化合物(1)(11.57g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(1.1g)、bafl(0.7g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰
亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0638]
《实施例i-12》
[0639]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(202g)、甲苯(20g)、作为二胺的daba(5.5g)、bisam(3.1g)、含硅化合物(2)(11.42g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(5.5g)、bisam(3.1g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0640]
《实施例i-13》
[0641]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(218g)、甲苯(22g)、作为二胺的daba(5.9g)、bafl(9.4g)、含硅化合物(1)(12.35g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(2.5g)、bafl(4.0g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0642]
《比较例i-1》
[0643]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表1和表2中记载的那样,边搅拌边添加作为溶剂的nmp(208g)、甲苯(21g)、作为二胺的daba(10.22g)、tfmb(16.0g),接着,在室温下添加作为酸二酐的cpoda(27.7g)、bpda(5.3g)、含硅化合物(6)(9.95g)。酸二酐与二胺的摩尔比为100:100。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温,得到聚酰亚胺树脂(除含硅化合物之外的官能团:酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。将所得清漆在冷冻库(设定为-20℃、以下同样)中保管,在进行评价时将其解冻而使用。
[0644]
《比较例i-2,i-3》
[0645]
在比较例i-1中,将溶剂、酸二酐、二胺的种类和量变更为表1和表2中记载的内容,除此之外,与比较例i-1同样地进行。
[0646]
《实施例i-14》
[0647]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(177g)、甲苯(18g)、作为二胺的含硅化合物(2)(4.12g)、33das(8.5g)、44das(12.8g),接着,在室温下添加作为酸二酐的odpa(31.0g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚
胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的33das(0.9g)、44das(1.4g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0648]
《实施例i-15》
[0649]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(170g)、甲苯(17g)、作为二胺的含硅化合物(2)(1.70g)、33das(8.7g)、44das(13.0g),接着,在室温下添加作为酸二酐的odpa(31.0g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的33das(1.0g)、44das(1.4g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0650]
《实施例i-16》
[0651]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(204g)、甲苯(20g)、作为二胺的含硅化合物(2)(16.36g)、daba(12.0g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(1.3g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0652]
《实施例i-17》
[0653]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(259g)、甲苯(26g)、作为二胺的含硅化合物(1)(15.54g)、bafl(29.1g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的bafl(3.2g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0654]
《实施例i-18》
[0655]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(232g)、甲苯(23g)、作为二胺的含硅化合物(1)(7.73g)、33das(8.5g)、44das(12.8g),接着,在室温下添加作为酸二酐的bpaf(45.8g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油
浴而恢复至室温。接着,边搅拌边添加作为二胺的33das(0.9g)、44das(1.4g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0656]
《实施例i-19》
[0657]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表3和表4中记载的那样,边搅拌边添加作为溶剂的nmp(235g)、甲苯(24g)、作为二胺的含硅化合物(1)(7.84g)、6foda(28.9g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的6foda(3.2g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。
[0658]
《比较例i-4》
[0659]
如表3和表4中记载的那样,将bpada 200g(0.384mol)分散至1,2-双(2-甲氧基乙氧基)乙烷(三甘醇二甲醚)1101g中,并保持至80℃。向其中投入含硅化合物x-22-9409(信越化学公司制、两个末端:氨基、官能团当量:670)172g(0.115mol),进行30分钟的均匀搅拌。接着,加热至140℃并进行1小时的搅拌,在结束反应后,升温至180℃,进行3小时的加热回流。在反应结束后,冷却至室温并投入水27.7g(1.54mol)。在均匀搅拌30分钟后,加热至80℃并进行3小时的加热回流。如此操作,得到溶解有经酰亚胺化的四羧酸(末端四羧酸硅氧烷酰亚胺低聚物)的溶液。
[0660]
接着,将溶液冷却至室温,投入mbaps 99.7g(0.230mol),在室温下进行1小时的均匀搅拌,得到树脂组合物溶液。
[0661]
《比较例i-5》
[0662]
如表3和表4中记载的那样,对三口可分离烧瓶安装氮气导入管、温度计、具备水分分离收集器的球形冷凝管。在室温25℃下投入三乙二醇二甲基醚(三甘醇二甲醚)15g、γ-丁内酯(gbl)35g、甲苯20.0g、odpa 10.86g(35.00mmol),搅拌至均匀。其后,升温至80℃,添加含硅化合物kf-8010(信越化学工业公司制、两个末端:氨基、官能团当量:430)11.30g(13.78mmol),进一步搅拌0.5小时后,升温至170℃,加热4小时。在反应中,副产的水与甲苯进行共沸,使用具备水分分离收集器的球形冷凝管,在回流下进行脱水。去除副产的水后,停止回流,将甲苯全部去除。将体系冷却至100℃后,添加马来酸酐0.14g,并搅拌0.5小时。
[0663]
在室温25℃下静置12小时,进行冷却后,添加apb 6.00g(20.52mmol),得到树脂组合物。
[0664]
《比较例i-6》
[0665]
如表3和表4中记载的那样,向具备不锈钢制半月型搅拌叶片、氮气导入管、安装有冷凝管的迪恩斯塔克、温度计、玻璃制管端盖板的500ml的5口圆底烧瓶中投入6foda 27.0g(0.0802摩尔)和nmp 56.000g,在体系内温度为70℃、氮气气氛下、转速为200rpm的条件下,进行搅拌而得到溶液。
[0666]
向该溶液中一并添加cpoda 19.2g(0.050摩尔)和nmp 14.000g后,投入作为酰亚
胺化催化剂的tea 0.253g,利用罩式加热器进行加热,耗时约20分钟将反应体系内的温度提升至190℃。捕集被馏去的成分,随着粘度上升而调整转速,且将反应体系内的温度保持至190℃,回流1小时。其后,添加nmp 85.806g,将反应体系内的温度冷却至50℃,得到包含具有酰亚胺重复结构单元的低聚物的溶液。
[0667]
向所得溶液中一并添加bpda 9.7g(0.033摩尔)和nmp 7.527g,以50℃搅拌5小时。其后,添加nmp 100.000g并进行均匀化后,投入在nmp 16.667g中溶解有x-22-1660b-3(改性硅油、官能团当量:2200、信越化学公司)13.2g(0.003摩尔)的混合液,进而搅拌约1小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。
[0668]
使用实施例和比较例的树脂组合物,进行粘度稳定性、过滤性的各评价,使用所得聚酰亚胺树脂膜,进行拉伸伸长率、残留应力的各评价。将评价结果示于表1~4。
[0669]
实施例和比较例中的简写如下所示。
[0670]
〈酸二酐〉
[0671]
cpoda:下述通式的化合物
[0672][0673]
odpa:4,4
’‑
氧双邻苯二甲酸酐
[0674]
6fda:4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐
[0675]
bpaf:9,9-双(3,4-二羧基苯基)芴二酸酐
[0676]
bzda:下述通式的化合物
[0677][0678]
bnbda:下述通式的化合物
[0679][0680]
bpada:2,2-双[4-(3,4-二羧基苯氧基)苯基]丙烷二酐
[0681]
〈二胺〉
[0682]
daba:3,5-二氨基苯甲酸
[0683]
3,3
’‑
二氨基二苯基砜(33das)
[0684]
4,4
’‑
二氨基二苯基砜(44das)
[0685]
bafl:9,9-双(4-氨基苯基)芴
[0686]
bisam:下述通式的化合物
[0687][0688]
tfmb:二氨基双(三氟甲基)联苯
[0689]
bapp:下述通式的化合物
[0690][0691]
mbaps:双[4-(3-氨基苯氧基)苯基]砜
[0692]
apb:1,3-双(3-氨基苯氧基)苯
[0693]
〈含硅化合物〉
[0694]
含硅化合物(1):(通式(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为1500的化合物)
[0695]
含硅化合物(2):(通式(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为800的化合物)
[0696]
含硅化合物(3):(通式(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为2200的化合物)
[0697]
含硅化合物(4):通式(10)中,l1和l2为氨基,r1为-ch2ch2ch
2-,r2、r3、r6、r7为甲基,r4、r5为苯基,j/(i+j+k)=0.15,官能团当量为2200的化合物
[0698]
含硅化合物(5):(通式(10)中,l1和l2为酸酐基(-ch(c=o)2o),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为1000的化合物)
[0699]
含硅化合物(6):(通式(10)中,l1和l2为酸酐基(-ch(c=o)2o),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为500的化合物)
[0700]
[表1]
[0701][0702]
[表2]
[0703][0704]
[表3]
[0705][0706]
[表4]
[0707][0708]
[表5]
[0709][0710]
〔第二方式〕
[0711]
《测定和评价方法》
[0712]
〈重均分子量〉
[0713]
重均分子量(mw)和数均分子量(mn)利用凝胶渗透色谱法(gpc),并利用下述条件来测定。
[0714]
作为溶剂,使用nmp(和光纯药工业公司制、高效液相色谱用、在即将测定之前添加24.8mmol/l的溴化锂一水合物(和光纯药工业公司制、纯度为99.5%)和63.2mmol/l的磷酸(和光纯药工业公司制、高效液相色谱用)并溶解而得到的物质)。用于计算重均分子量的标准曲线使用标准聚苯乙烯(东曹公司制)来制作。
[0715]
柱:shodex kd-806m(昭和电工公司制)
[0716]
流速:1.0ml/分钟
[0717]
柱温度:40℃
[0718]
泵:pu-2080plus(jasco公司制)
[0719]
检测器:ri-2031plus(ri:差示折射计、jasco公司制)和uv-2075plus(uv-vis:紫外可见吸光度计、jasco公司制)
[0720]
《组合物(清漆)的粘度稳定性的评价》
[0721]
将实施例和比较例中制备的树脂组合物在23℃、50%rh的气氛下放置24小时,使用带有调温机的粘度计(东机产业公司制的viscomater tve-35h),测定树脂组合物的初始(0天)的粘度。并且,将树脂组合物在23℃、50%rh的气氛下放置7天,与初始同样操作,测定7天后的粘度。接着,计算下述式所示的粘度稳定性。
[0722]
粘度稳定性(%/day)=|(初始粘度(mpa
·
s))-(7天后的粘度(mpa
·
s))|/(初始粘度(mpa
·
s))
×
100/7(day)
[0723]
粘度稳定性按照下述基准进行评价。
[0724]
a:粘度稳定性为1%/day以下“优良”[0725]
b:粘度稳定性超过1%/day且为3%/day以下“良”[0726]
c:粘度稳定性超过3%/day且为5%/day以下“可”[0727]
d:粘度稳定性超过5%/day“可”[0728]
《组合物(清漆)的过滤性的评价》
[0729]
将实施例和比较例中制备的树脂组合物在23℃、50%rh的气氛下,使用尺寸为900mmφ、单位面积重量为0.2μm的聚乙烯制的薄膜过滤器,以0.25mpa的压力进行加压过滤。测定此时的过滤出的树脂组合物的质量,计算过滤速度。过滤性按照下述基准进行评价。
[0730]
a:过滤速度为2000g/hr以上“优”[0731]
b:过滤速度小于2000g/hr且为1500g/hr以上“良”[0732]
c:过滤速度小于1500g/hr且为1000g/hr以上“可”[0733]
d:过滤速度小于1000g/hr“不可”[0734]
《聚酰亚胺树脂膜的拉伸伸长率的评价》
[0735]
作为支承体,使用在表面设置有铝蒸镀层的6英寸硅晶圆基板,在该铝蒸镀层的面上以聚酰亚胺树脂膜的膜厚成为10μm的方式旋涂实施例和比较例中制备的树脂组合物,形成涂膜。将该涂膜以100℃预烘烤6分钟后,使用库内的氧浓度被调整至10质量ppm以下的立式固化炉(koyo thermo system公司制、型号名“vf-2000b”),以400℃加热1小时,在晶圆上
形成聚酰亚胺薄膜。接着,使用切割锯(disco公司制、品名“dad3350”),对所得聚酰亚胺薄膜划入3mm宽的划痕后,将带有聚酰亚胺薄膜的晶圆在稀盐酸水溶液中浸渍一晚,剥离聚酰亚胺薄膜片并使其干燥,得到3mm宽的聚酰亚胺片。将其切割成50mm长,得到3mm宽、50mm长的聚酰亚胺测定试样。使用tensilon(orientec公司制、型号名“utm-ii-20”),在试验速度为40mm/分钟、初始载荷为0.5fs的条件下,测定聚酰亚胺树脂膜的拉伸伸长率。拉伸伸长率按照下述基准进行评价。
[0736]
a:拉伸伸长率超过50%“优良”[0737]
b:拉伸伸长率超过40%且为50%以下“良”[0738]
c:拉伸伸长率超过30%且为40%以下“可”[0739]
d:拉伸伸长率小于30%“不可”[0740]
《聚酰亚胺树脂膜的残留应力的评价》
[0741]
利用旋涂机,在预先测定过“翘曲量”的厚度625μm
±
25μm的6英寸硅晶圆上涂布实施例和比较例中制备的树脂组合物,在100℃下预烘烤7分钟。其后,使用立式固化炉(koyo lindberg公司制、型号名:vf-2000b),以库内的氧浓度成为10质量ppm以下的方式进行调整,在400℃下实施1小时的加热固化处理(固化处理),制作在固化后带有膜厚10μm的聚酰亚胺树脂膜的硅晶圆。
[0742]
使用残留应力测定装置(tencor公司制、型号名:flx-2320)来测定该晶圆的翘曲量,评价“在氮气气氛下”在硅晶圆与树脂膜之间产生的残留应力。
[0743]
a:残留应力小于30mpa“良”[0744]
b:残留应力为30mpa以上且小于40mpa“可”[0745]
c:拉伸伸长率为40mpa以上“不可”[0746]
《实施例ii-1》
[0747]
在带有搅拌棒的3l可分离烧瓶中,边导入氮气,边如表6和表7中记载的那样,边搅拌边添加作为溶剂的nmp(191g)、作为二胺的daba(14.4g)和含硅化合物(1)(10.82g),接着,添加作为酸二酐的cpoda(38.4g)。酸二酐与二胺的摩尔比为100:98。将混合物在室温下搅拌48小时,得到透明的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。将所得清漆在冷冻库(设定为-20℃、以下同样)中保管,在进行评价时将其解冻而使用。
[0748]
《实施例ii-2》
[0749]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表6和表7中记载的那样,边搅拌边添加作为溶剂的nmp(191g)、甲苯(19g)、作为二胺的daba(14.4g)和含硅化合物(1)(10.82g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。酸二酐与二胺的摩尔比为100:98。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温,得到聚酰亚胺树脂(除含硅化合物之外的官能团:酰亚胺基;含硅化合物的官能团:酰亚胺基)的nmp溶液。将所得清漆在冷冻库(设定为-20℃、以下同样)中保管,在进行评价时将其解冻而使用。
[0750]
《实施例ii-3》
[0751]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮
气,边如表6~表9中任一者中记载的那样,边搅拌边添加作为溶剂的nmp(189g)、甲苯(19g)、作为二胺的daba(13.9g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加含硅化合物(2)(10.72g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。
[0752]
《实施例ii-16》
[0753]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表8和表9中记载的那样,边搅拌边添加作为溶剂的nmp(177g)、甲苯(18g)、作为二胺的33das(8.5g)和44das(12.8g),接着,在室温下添加作为酸二酐的odpa(31.0g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的33das(0.9g)和44das(1.4g)、以及含硅化合物(2)(4.12g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。
[0754]
《实施例ii-17》
[0755]
向在上部具备迪恩斯塔克管和回流管且带有搅拌棒的可分离烧瓶中,边导入氮气,边如表8和表9中记载的那样,边搅拌边添加作为溶剂的nmp(173g)、甲苯(17g)、作为二胺的daba(13.0g),接着,在室温下添加作为酸二酐的cpoda(38.4g)。其后,将内部温度升温至160℃,以160℃进行1小时的加热回流,进行酰亚胺化。在酰亚胺化结束后,升温至180℃,边取出甲苯边持续反应。在反应12小时后,卸除油浴而恢复至室温。接着,边搅拌边添加作为二胺的daba(1.4g)和含硅化合物(2)(4.80g)。酸二酐与二胺的摩尔比为100:98。并且,将混合物在室温下搅拌48小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。
[0756]
《比较例ii-4》
[0757]
如表8和9中记载的那样,向具备不锈钢制半月型搅拌叶片、氮气导入管、安装有冷凝管的迪恩斯塔克、温度计、玻璃制管端盖板的500ml的5口圆底烧瓶中投入6foda 27.0g(0.0802摩尔)和nmp 56.000g,在体系内温度为70℃、氮气气氛下、转速为200rpm的条件下,进行搅拌而得到溶液。
[0758]
向该溶液中一并添加cpoda 19.2g(0.050摩尔)和nmp 14.000g后,投入作为酰亚胺化催化剂的tea0.253g,利用罩式加热器进行加热,耗时约20分钟将反应体系内的温度提升至190℃。捕集被馏去的成分,随着粘度上升而调整转速,并且,将反应体系内的温度保持至190℃,回流1小时。其后,添加nmp 85.806g,将反应体系内的温度冷却至50℃,得到包含具有酰亚胺重复结构单元的低聚物的溶液。
[0759]
向所得溶液中一并添加bpda 9.7g(0.033摩尔)和nmp 7.527g,以50℃搅拌5小时。其后,添加nmp 100.000g并进行均匀化后,投入在nmp 16.667g中溶解有x-22-1660b-3(改
性硅油、官能团当量:2200、信越化学)13.2g(0.003摩尔)的混合液,进一步搅拌约1小时,得到部分进行了酰亚胺化的聚酰亚胺前体(除含硅化合物之外的官能团:酰胺基或酰亚胺基;含硅化合物的官能团:酰胺基)的nmp溶液(以下也称为清漆)。
[0760]
《实施例ii-4、ii-5、ii-7、ii-9、ii-10、ii-12、ii-14、ii-15》
[0761]
在实施例ii-3中,将溶剂、酸二酐、二胺、含硅化合物的种类和量变更为表6和表7中记载的内容,除此之外,与实施例ii-3同样地进行。
[0762]
《参考例ii-6、实施例ii-8、ii-11、ii-13、比较例ii-1~ii-3》
[0763]
在实施例ii-2中,将溶剂、酸二酐、二胺、含硅化合物的种类和量变更为表6和表7中记载的内容,除此之外,与实施例ii-2同样地进行。
[0764]
使用实施例和比较例的树脂组合物,进行粘度稳定性、过滤性的各评价,使用所得聚酰亚胺树脂膜,进行拉伸伸长率、残留应力的各评价。将评价结果示于表6~表9。
[0765]
实施例和比较例中的简写如下所示。
[0766]
〈酸二酐〉
[0767]
cpoda:下述通式的化合物
[0768][0769]
odpa:4,4
’‑
氧双邻苯二甲酸酐
[0770]
6fda:4,4
’‑
(六氟异亚丙基)双邻苯二甲酸酐
[0771]
bpaf:9,9-双(3,4-二羧基苯基)芴二酸酐
[0772]
bzda:下述通式的化合物
[0773][0774]
bnbda:下述通式的化合物
[0775][0776]
〈二胺〉
[0777]
daba:3,5-二氨基苯甲酸
[0778]
bafl:9,9-双(4-氨基苯基)芴
[0779]
bisam:下述通式的化合物
[0780][0781]
tfmb:二氨基双(三氟甲基)联苯
[0782]
bapp:下述通式的化合物
[0783][0784]
33das:3,3
’‑
二氨基二苯基砜
[0785]
44das:3,3
’‑
二氨基二苯基砜
[0786]
6foda:2,2
’‑
双(三氟甲基)-4,4
’‑
二氨基二苯基醚
[0787]
〈含硅化合物〉
[0788]
含硅化合物(1):(通式(5)、(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为1500的化合物)
[0789]
含硅化合物(2):(通式(5)、(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为800的化合物)
[0790]
含硅化合物(3):(通式(5)、(10)中,l1和l2为氨基(-nh2),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为2200的化合物)
[0791]
含硅化合物(4):通式(5)、(10)中,l1和l2为氨基,r1为-ch2ch2ch
2-,r2、r3、r6、r7为甲基,r4、r5为苯基,j/(i+j+k)=0.15,官能团当量为2200的化合物
[0792]
含硅化合物(5):(通式(5)、(10)中,l1和l2为酸酐基(-ch(c=o)2o),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为1000的化合物)
[0793]
含硅化合物(6):(通式(5)、(10)中,l1和l2为酸酐基(-ch(c=o)2o),r1为三亚甲基(-ch2ch2ch
2-),r2、r3为甲基,j、k为0,官能团当量为500的化合物)
[0794]
[表6]
[0795][0796]
[表7]
[0797][0798]
[表8]
[0799][0800]
[表9]
[0801][0802]
附图标记说明
[0803]
2a
ꢀꢀ
下部基板
[0804]
2b
ꢀꢀ
密封基板
[0805]
25
ꢀꢀ
有机el结构部
[0806]
250a
ꢀꢀ
发出红色光的有机el元件
[0807]
250b
ꢀꢀ
发出绿色光的有机el元件
[0808]
250c
ꢀꢀ
发出蓝色光的有机el元件
[0809]
251
ꢀꢀ
分隔壁(堤)
[0810]
252
ꢀꢀ
下部电极(阳极)
[0811]
253
ꢀꢀ
空穴传输层
[0812]
254
ꢀꢀ
发光层
[0813]
255
ꢀꢀ
上部电极(阴极)
[0814]
256
ꢀꢀ
tft
[0815]
257
ꢀꢀ
接触孔
[0816]
258
ꢀꢀ
层间绝缘膜
[0817]
259
ꢀꢀ
下部电极
[0818]
261
ꢀꢀ
中空部。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1