一种溴化苄的制备方法与流程
1.本技术涉及甲基苯类化合物溴代做卞溴一类的有机合成反应技术领域。尤其涉及一种溴化苄的制备方法。
背景技术:
2.甲基苯类化合物用nbs、二溴海因等溴代试剂直接溴代为卞溴,是常用的比较直接的做溴化苄的方式。此过程会存在原料残留,二溴代,甚至三溴代的杂质,这些杂质与产品极性差别不大,沸点类似,各方面性质很接近,再加上卞溴类化合物自身会比较刺激,所以纯化起来比较繁琐且容易引起人不适感。
3.目前,通常会引入aibn、bpo等引发剂,使得反应温度,底物浓度等控制在合适的最佳状态,但是不可避免的会有二溴代,原料残余等情况存在。然后,再通过精馏、过柱子、重结晶等方式纯化,操作比较繁琐不易,想把产品纯度搞上去不容易。
技术实现要素:
4.本技术提供一种溴化苄的制备方法,用以解决采用现有技术提供的溴化苄制备方法制得的溴化苄纯度低,且纯化过程复杂的技术问题。具体方案如下:
5.本技术提供给了一种溴化苄的制备方法,包括:
6.将甲苯类化合物加入溶剂中形成混合物;
7.在所述混合物中加入溴代试剂以及引发剂,升温至引发反应,得到第一溶液;
8.去除所述第一溶液中的滤渣,得到滤液,并在滤液中滴入碱以及掉溴试剂,去除所述滤液中的二溴代物,得到第二溶液;
9.对所述第二溶液进行萃取、干燥,得到目标物;
10.对所述目标物进行减压浓缩处理,并去除处理后的目标物中的溶剂,得粗品溴化苄;
11.对所述粗品溴化苄进行重结晶,得到溴化苄。
12.可选的,所述溶剂为dmf,乙腈,二氯甲烷,二氯乙烷,四氯化碳中的任一种。
13.可选的,所述将甲苯类化合物加入溶剂中形成混合物包括:
14.反应釜抽入所述溶剂,并搅拌;
15.将所述甲苯类化合物加入所述反应釜中,搅拌以形成所述混合物。
16.可选的,在所述混合物中加入溴代试剂以及引发剂,并升温至引发进行反应,得到第一溶液包括:
17.在所述混合物中加入第一批溴代试剂以及引发剂并升温至引发进行反应,得到预处理溶液;
18.在所述预处理溶液中加入第二批溴代试剂以及引发剂并升温至引发进行反应,得到第一溶液。
19.可选的,在所述混合物中加入溴代试剂以及引发剂,并升温至引发进行反应,得到
第一溶液中,所述回流反应的终止标准为:
20.按浓度百分比计,所述第一溶液中的甲苯类化合物小于5%。
21.可选的,所述溴代试剂为n-溴代丁二酰亚胺或二溴海因。
22.可选的,所述引发剂为偶氮二异丁腈或过氧化二苯甲酰。
23.可选的,所述对所述第二溶液进行萃取、干燥,得到目标物之前,所述方法还包括:
24.调节所述第二溶液的ph值至4-5。
25.可选的,所述碱为三乙胺或n,n-二异丙基乙胺。
26.可选的,所述掉溴试剂为亚磷酸二乙酯或亚磷酸二甲酯。
27.本技术提供的溴化苄的制备方法,包括:将2-氰基-5-氟甲苯加入溶剂中形成混合物;在所述混合物中加入溴代试剂以及引发剂,并升温至85-90℃进行回流反应,得到第一溶液;去除所述第一溶液中的滤渣,得到滤液,并在滤液中滴入碱以及掉溴试剂,去除所述滤液中的二溴代物,得到第二溶液;对所述第二溶液进行萃取、干燥,得到目标物;对所述目标物进行减压浓缩处理,并去除处理后的目标物中的溶剂,得粗品溴化苄;对所述粗品溴化苄进行重结晶,得到溴化苄。本技术通过增加掉溴过程,可以提高原料的转化率,并且避免纯化二溴代,使得固体产物不用精馏,简化了溴化苄的纯化过程。
附图说明
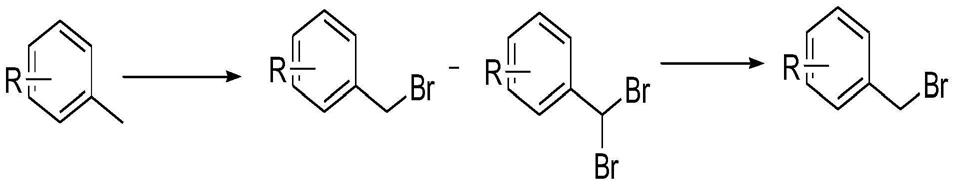
28.图1是本发明提供的一种溴化苄的反应过程图。
具体实施方式
29.为使本技术实施例的目的、技术方案和优点更加清楚,下面将结合本技术实施例中的附图,对本技术实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本技术一部分实施例,而不是全部的实施例。基于本技术中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本技术保护的范围。
30.在本技术实施例的描述中,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本技术的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。在本技术中,除非另有明确的规定和限定,术语“安装”、“相连”、“连接”、“固定”等术语应做广义理解,例如,可以是固定连接,也可以是可拆卸连接,或成一体;可以是直接相连,也可以通过中间媒介间接相连,可以是两个元件内部的连通或两个元件的相互作用关系,除非另有明确的限定。对于本领域的普通技术人员而言,可以根据具体情况理解上述术语在本技术中的具体含义。
31.在本技术中,除非另有明确的规定和限定,第一特征在第二特征“上”或“下”可以是第一和第二特征直接接触,或第一和第二特征通过中间媒介间接接触。而且,第一特征在第二特征“之上”、“上方”和“上面”可是第一特征在第二特征正上方或斜上方,或仅仅表示第一特征水平高度高于第二特征。第一特征在第二特征“之下”、“下方”和“下面”可以是第一特征在第二特征正下方或斜下方,或仅仅表示第一特征水平高度小于第二特征。
32.在本技术的描述中,需要理解的是,术语“内”、“外”、“上”、“底”、“前”、“后”等指示的方位或者位置关系(若有的话)为基于附图1所示的方位或位置关系,仅是为了便于描述
本技术和简化描述,而不是指示或者暗示所指的装置或者元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本技术的限制。
33.实施例1
34.本技术提供给了一种溴化苄的制备方法,包括:
35.步骤s101,将甲苯类化合物加入溶剂中形成混合物。
36.具体的,上述甲苯类化合物可以是2-氰基-5-氟甲苯,也可以是其它甲苯类化合物,具体到本技术中,可以将任意的甲苯类化合物作为本技术的原料,进行溴化苄的制备,本技术不对上述甲苯类化合物的种类做限定。
37.以2-氰基-5-氟甲苯为例制备溴化苄,具体的,可以釜中抽入溶剂,开启搅拌;接着向釜中加入2-氰基-5-氟甲苯,以形成混合物,需要说明的是,上述溶剂可以是dmf(n,n-dimethylformamide,n,n-二甲基甲酰胺),乙腈,二氯甲烷,二氯乙烷,四氯化碳中的任一种。
38.具体的,上述2-氰基-5-氟甲苯还可以用nbs(n-bromosuccinimide,n-溴代丁二酰亚胺)替代。底物nbs一般在1.05-1.5当量,二溴海因会在0.55-0.9当量。
39.步骤s102,在所述混合物中加入溴代试剂以及引发剂,并升温至85-90℃进行回流反应,得到第一溶液。
40.具体的,所述溴代试剂为n-溴代丁二酰亚胺或二溴海因;上述引发剂为偶氮二异丁腈或过氧化二苯甲酰,作为优选的方式,采用过氧化二苯甲酰作为引发剂,器引发温和,稳定性好,aibn引发猛烈,高温易分解。
41.具体的,上述溴代试剂以及引发剂可以分为两批次加入到混合物中,例如,第一次加入预定量的溴代试剂以及引发剂,升温至85-90℃进行回流反应,接着,降温至60-70℃,在加入第二批次的溴代试剂以及引发剂,继续升温至85-90℃进行回流反应,以得到第一溶液,为了确保成品的纯度,可以对上述第一溶液中原路(即2-氰基-5-氟甲苯)的含量做限定,例如,按浓度百分比计,所述第一溶液中的2-氰基-5-氟甲苯小于5%。
42.步骤s103,去除所述第一溶液中的滤渣,得到滤液,并在滤液中滴入碱以及掉溴试剂,去除所述滤液中的二溴代物,得到第二溶液。
43.具体的,上述碱为三乙胺或n,n-二异丙基乙胺;上述掉溴试剂为亚磷酸二乙酯或亚磷酸二甲酯。
44.具体的,可以将第一溶液的反应降温至20℃以下,对第一溶液过滤,滤饼100kg二氯乙烷淋洗,滤渣弃去,以得到滤液。滤液合并加入釜中,降温至0℃以下,滴加亚磷酸二乙酯8.5kg,30min滴完,控温-5~5℃,滴三乙胺7.2kg,3h滴完,保持-5~5℃,滴完保温反应30min,取样hplc/gc检测,二溴代反应完全,终止反应。
45.步骤s104,对所述第二溶液进行萃取、干燥,得到目标物。
46.具体的,加水100kg,加盐酸调ph至4-5,分液,水相50kg二氯乙烷萃取一次,,分液,水相弃去,有机相合水50kg洗,水相弃去,有机相用11kg无水硫酸钠干燥,得到目标物。
47.步骤s105,对所述目标物进行减压浓缩处理,并去除处理后的目标物中的溶剂,得粗品溴化苄。
48.具体的,有机相55-65℃,减压浓缩至无溶剂,加入石油醚,加热至50-60℃溶解,放料,降至10度,过滤,得粗品溴化苄。
49.步骤s106,对所述粗品溴化苄进行重结晶,得到溴化苄。
50.具体的,上述重结晶的过程可以是釜中加入正庚烷120kg,加入61kg物料,升温至80℃,回流2h,观察料完全溶解后,降温至10-20℃析晶,离心得58kg,石油醚淋洗30kg;得到的溴化苄经检测:gc≧99.5%,单杂0.25%。
51.本技术通过增加掉溴过程,可以提高原料的转化率,并且避免纯化二溴代,使得固体产物不用精馏,简化了溴化苄的纯化过程。
52.实施例2
53.在上述实施例1的基础上,本方案以制备58kg溴化苄为例,对本技术的技术方案做详细阐述,具体入下:
54.步骤1,釜中抽入165kg二氯乙烷,开启搅拌;向釜中加入26.5kg 2-氰基-5-氟甲苯,二溴海因19.05kg,aibn(azodiisobutyronitrile;azobisisobutyronitrile,偶氮二异丁腈)1.2kg,升温至80-85℃,回流反应1h,降温至60-70℃,第二次加二溴海因19.05kg,aibn1.2kg,继续反应1h,hplc检测,原料小于5%终止反应。
55.步骤2,反应降温至20℃以下,过滤,滤饼100kg二氯乙烷淋洗,滤渣弃去。
56.步骤3,滤液合并加入釜中,降温至0℃以下,滴加亚磷酸二乙酯8.5kg,30min滴完,控温-5~5℃,滴三乙胺7.2kg,3h滴完,保持-5~5℃,滴完保温反应30min,取样hplc/gc检测,二溴代反应完全,终止反应。
57.步骤4,加水100kg,加盐酸调ph至4-5,分液,水相50kg二氯乙烷萃取一次,,分液,水相弃去,有机相合水50kg洗,水相弃去,有机相用11kg无水硫酸钠干燥。
58.步骤5,有机相55-65℃,减压浓缩至无溶剂,加入石油醚35kg,加热至50-60℃溶解,放料,降至10度,过滤,得粗品79kg。
59.步骤6,重结晶:釜中加入正庚烷120kg,加入61kg物料,升温至80℃,回流2h,观察料完全溶解后,降温至10-20℃析晶,离心得58kg,石油醚淋洗30kg,gc≧99.5%,单杂0.25%。
60.对比例
61.现有手段:在溴化苄的制备领域中,目前的制备方法为:2-氰基-5-氟溴苄(cas:421552-12-7)为曲格列汀的关键起始原料,市场现在是用乙腈作溶剂,二溴海因溴化试剂,bpo引发剂,在密闭条件下,升温85-90度,得到产品原料剩余15%,产物75%,二溴10%,最终产品,但是,上述方法中,得到的产品需要装2.5m高的精馏柱纯化产品,得到产物后,再用正庚烷重结晶,得到合格样品;为了确保产品的合格率,现有技术手段只能通过反复重结晶或者用柱效很高的精馏柱去精馏纯化,操作繁琐。纯化成本高,过程复杂,且纯化效果差。
62.申请人对上述现有技术的成品进行检测,其检测结果如下:
63.乙腈闷罐升温85-90度,得到产品原料剩余15%,产物75%,二溴10%。
64.申请人对本方案提供的溴化苄的制备方法制得的溴化苄进行检测,其检测结果如下:
65.常规反应釜,80-85度,原料剩余1%,产物92%,二溴2%。
66.可见,采用本实施例提供的溴化苄的制备方法,反应过程中,增大溴代试剂的用量,使得原料最大限度降到最低,然后,根据二溴代含量,加入掉溴试剂,使得二溴杂质转化为产品,从而提高产品转化率。在制备过程中,存在掉溴过程,所以转化率提高,故收率也大
大提高;采用本实施例的方法制得的粗品杂质少,易纯化;采用本实施例的方法制备溴化苄,收率提高,易纯化,成本低;对于不易反应的底物,避免闷起来反应,安全性增加。
67.以上,仅为本技术的具体实施方式,但本技术的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本技术揭露的技术范围内,可轻易想到变化或替换,都应涵盖在本技术的保护范围之内。因此,本技术的保护范围应以权利要求的保护范围为准。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1