一种精制氮丙啶的制备方法与流程
1.本发明属于化工领域,具体涉及一种新型的精制氮丙啶制备方法。
背景技术:
2.氮丙啶是一种反应能力很强的三元环化合物,可与带有多种官能团的化合物反应,所发生的反应主要有取代反应、开环反应和共聚反应。经开环聚合或共聚可得到反应性高分子化合物,在造纸工业中,粘合剂合成,在纺织工业中,化妆品工业等领域都有广泛应用。而氮丙啶单体同样可用于制造染料、农药、数码照相激光冲印感光材料、氮丙啶交联剂等。目前氮丙啶单体的合成方法主要有硫酸酯化法,2-氯乙胺法,二氯乙烷法,环氧乙烷法和烷烃胺气相脱水法;德国巴斯夫主要采用一乙醇胺液相法,而日本触媒采用比较先进的一乙醇胺气相法,工艺简单,但催化剂是核心技术,目前还没有其它公司采用一乙醇胺气相法,国内目前工业化主要采用的一乙醇胺硫酸酯化法;目前氮丙啶的提纯都采用精馏的方法,由于现有技术合成出的氮丙啶含水量高(大约50%-80%),副产物多,减压精馏不仅耗时耗力,单次精馏氮丙啶纯度不高,只能多次重复精馏,不然很难满足精细化工,医药,氮丙啶交联剂等方面需求。
3.专利申请号为:201910296900.8中粗品氮丙啶的提纯需要在45-95℃下,蒸馏4-10小时,重复3-5次,然后减压条件下收集20-60℃的馏分8-12小时;专利申请号为:201910196848.9公开了一种制备甲基氮丙啶的方法,其中甲基氮丙啶的精制工艺和申请号为:201910296900.8的专利类似,提纯产物的方法耗时耗能,并且得到的氮丙啶纯度不够,水分含量过高。
技术实现要素:
4.针对现有技术的不足,本发明的目的在于提供一种新型的精制氮丙啶制备方法。本发明采用分步精制的方法;第一步通过简单的蒸馏将盐,高沸点副产物,未反应原料和氮丙啶水溶液分离出来;第二步氮丙啶水溶液通入含氢氧化钠的混合釜中,在0-10℃使溶液达到饱和,由于氮丙啶难溶于氢氧化钠饱和溶液,静置后分层,上层氮丙啶含量可以达到90%以上;第三步将上层的氮丙啶通过装有填料的精馏塔中,单次精馏后的纯度达到99%以上,完全满足应用需求。
5.为实现上述目的,本发明采用以下技术方案:
6.一种精制氮丙啶的制备方法:包括以下步骤:
7.(1)在0℃-10℃氮丙啶粗品与氢氧化钠加入反应釜中并使氢氧化钠在体系中饱和,搅拌后静置;
8.(2)将上层含氮丙啶的油相与下层氢氧化钠水溶液分离;
9.(3)对步骤(2)中分离出来的氮丙啶进行精馏。
10.在上述制备方法中,作为一种优选实施方式,步骤(1)中,静置温度为0℃-10℃(例如1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃);优选地,搅拌时间为1h-3h(例如1.5h、2h、
2.5h);优选地,静置时间为1h-3h(例如1.5h、2h、2.5h)。
11.在上述制备方法中,作为一种优选实施方式,步骤(1)中将氮丙啶粗品与氢氧化钠加入反应釜中并使氢氧化钠在体系中饱和的操作包括:称取氮丙啶粗品,加入装有氢氧化钠的0℃-10℃(例如1℃、2℃、3℃、4℃、5℃、6℃、7℃、8℃、9℃)的反应釜中,然后开动搅拌,继续加氢氧化钠,直至体系中氢氧化钠达到过饱和;优选地,所述氢氧化钠为片状氢氧化钠固体即片碱。
12.在上述制备方法中,作为一种优选实施方式,所述氮丙啶粗品为5wt%-60wt%(例如10wt%、20wt%、30wt%、40wt%、50wt%)的氮丙啶水溶液,优选为40wt-48wt%(例如42wt%、45wt%、46wt%)的氮丙啶水溶液。
13.本发明中,由于氮丙啶难溶于氢氧化钠饱和溶液,包含氮丙啶的油相与下层氢氧化钠水溶液静置后分层,包含氮丙啶的油相进一步精馏提纯,下层水溶液可以配置成30%-50%水溶液循环使用于氮丙啶粗品的制备。
14.在上述制备方法中,作为一种优选实施方式,步骤(3)中的精馏在保护气氛下进行;进一步优选地,所述保护气氛为氮气。
15.在上述制备方法中,作为一种优选实施方式,步骤(3)中的精馏为单次精馏;优选地,所述精馏是在减压条件下完成的;优选地,步骤(3)中所述精馏温度为30℃-50℃(例如35℃、40℃、45℃),压力为0.6mpa-08mpa,时间为3h-7h(例如4h、5h、6h)。
16.在上述制备方法中,作为一种优选实施方式,所述精馏采用精馏釜完成。
17.本发明的所述精馏釜包括釜体,精馏塔和树脂填料;进一步优选地,所述精馏塔高度为3-6米。
18.在上述制备方法中,作为一种优选实施方式,步骤(1)中的氮丙啶粗品的制备方法为硫酸酯化法;更优选地,所述氮丙啶粗品的制备方法包括以下步骤:(a)在醇胺中滴加强酸,滴毕保温反应,保温结束后对反应体系进行减压蒸馏;(b)步骤(a)的减压蒸馏产物降温后,加入氢氧化钠水溶液,保温反应;(c)对步骤(b)的反应体系进行减压蒸馏,得到氮丙啶水溶液粗品。
19.本发明的制备氮丙啶粗品的方法无需加入催化剂即可进行。
20.在上述制备方法中,作为一种优选实施方式,所述减压蒸馏的压力为0.1mpa-0.9mpa(例如0.2mpa、0.3mpa、0.4mpa、0.5mpa、0.6mpa、0.7mpa、0.8mpa),温度为30℃-100℃(例如40℃、50℃、60℃、70℃、80℃、90℃)。
21.在上述制备方法中,作为一种优选实施方式,以氮丙啶粗品制备的原料醇胺、强酸和氢氧化钠水溶液的总重量来计算,其中所述醇胺的用量为10wt%-50wt%(例如20wt%、30wt%、40wt%),所述强酸的用量为10wt%-35wt%(例如15wt%、20wt%、25wt%),氢氧化钠水溶液的用量为30wt%-70wt%(例如40wt%、50wt%、60wt%);进一步优选地,所述氢氧化钠水溶液为35wt%的氢氧化钠水溶液。
22.在上述制备方法中,作为一种优选实施方式,步骤(a)中保温温度为70℃-90℃(例如75℃、80℃、85℃),保温时间为2-5小时(例如2.5h、3h、3.5h、4h、4.5h);优选地,步骤(b)中降温是指降温至20℃-30℃;优选地,步骤(b)中保温温度为30℃-50℃(例如35℃、40℃、45℃),进一步优选为45℃;优选地,步骤(b)中保温时间为2-5小时(例如2.5h、3h、3.5h、4h、4.5h),进一步优选为3小时。
23.在上述制备方法中,作为一种优选实施方式,所述强酸为浓硫酸或浓盐酸。
24.在上述制备方法中,作为一种优选实施方式,所述醇胺为单乙醇胺。
25.在上述制备方法中,作为一种优选实施方式,当所述单乙醇胺替换为异丙醇胺时,得到的产物氮丙啶被替换为甲基氮丙啶。
26.一种精制氮丙啶,所述精制氮丙啶由上述制备方法制备得到,优选地,所述精制氮丙啶中氮丙啶的重量百分比大于99%,进一步优选地,所述精制氮丙啶水分的重量百分比小于0.02%。
27.与现有技术相比,本发明的有益效果是:本发明所精制的氮丙啶单体用气相法测纯度99%以上,水分含量在0.02%以下,所得产品为无色透明液体;本发明中在0-10℃下氢氧化钠的溶解度为100g水中溶解40-50g氢氧化钠,而且在0-10℃氮丙啶在氢氧化钠饱和溶液的溶解度最低,氮丙啶的收率最大,需要的氢氧化钠最少,降低成本;本产品中用于提纯粗品氮丙啶的饱和碱溶液可以循环使用,节能环保,并且降低了成本;通过本发明精制的氮丙啶与三羟丙烷三丙烯酸酯发生迈克尔加成反应得到氮丙啶交联剂,与同类产品相比,检测指标和应用性能达到或超过同类产品。
附图说明
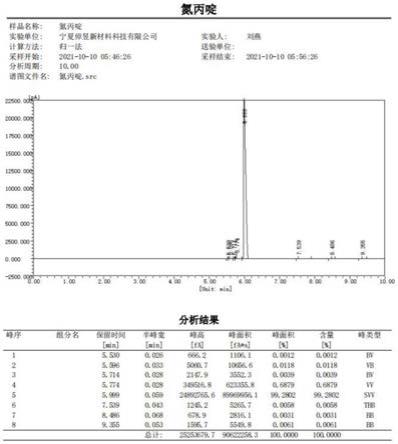
28.图1为实施例1中精制氮丙啶的气相检测结果;
29.图2为实施例2中精制2-甲基氮丙啶的气相检测结果;
30.图3为实施例3中精制氮丙啶的气相检测结果;
31.图4为对比例1中制备的氮丙啶的气相检测结果。
具体实施方式
32.下面结合具体实施方式对本发明精制氮丙啶制备方法进行进一步的详细描述,给出的实施例仅为了阐明本发明,而不是为了限制本发明的范围。以下提供的实施例可作为本技术领域普通技术人员进行进一步改进或应用的基础,并不以任何方式构成对本发明的具体限制。
33.以下实施例中使用的各种试剂和原料均为市售产品,单乙醇胺和2-甲基乙醇胺的纯度均为99%以上。
34.实施例1
35.粗氮丙啶制备:准确称取67g单乙醇胺,常温滴加98%浓硫酸108g,4h滴完,滴毕保温温度为70℃,保温3h,90℃、0.5mpa进行减压蒸馏,3h蒸完,降温至30℃,加入质量浓度为35%的276g氢氧化钠水溶液,升温40℃,反应3h,50℃、0.8mpa进行减压蒸馏5h,得到含量为40%氮丙啶水溶液138g。
36.精制氮丙啶制备:取含量为40%氮丙啶水溶液138g,加入61g氢氧化钠于温度为5℃的反应釜中,开动搅拌,使氢氧化钠溶液达到饱和状态,未饱和时继续加30g片碱,直至反应溶液达到饱和为止,搅拌2h,静置3h,静置温度为5℃,静置结束后将上层氮丙啶转移精馏釜,通氮气,进行精馏,精馏条件为:0.8mpa,50℃,精馏得到纯度大于99%的氮丙啶单体53g。下层氢氧化钠水溶液配置成30-50%水溶液可以循环使用于粗氮丙啶的制备。
37.对本实施例所制备的氮丙啶单体进行气相色谱纯度检测,检测结果如附图1和下
表1所示。
38.表1
39.项目名称纯度/%水分/%精制氮丙啶99.280.01
40.实施例2
41.粗2-甲基粗氮丙啶制备:准确称取67g异丙醇胺醇胺,常温滴加98%浓硫酸120g,4h滴完,滴毕保温温度为70℃,保温3h,90℃、0.5mpa进行减压蒸馏,3h蒸完,降温至30℃,加入质量浓度为35%的205g氢氧化钠水溶液,升温40℃,反应3h,50℃、0.8mpa进行减压蒸馏5h,得到含量为48%甲基氮丙啶水溶液158g。
42.精制氮丙啶制备:取含量为48%甲基氮丙啶水溶液158g,加入80g氢氧化钠于温度为5℃的反应釜中,开动搅拌,使氢氧化钠溶液达到饱和状态,未饱和时继续加10g片碱,直至反应溶液达到饱和为止,搅拌2h,静置3h,静置温度为5℃,静置结束后将上层2-甲基氮丙啶转移精馏釜,通氮气,进行精馏,精馏条件为:60℃,0.8mpa,精馏得到纯度大于99%的甲基氮丙啶单体73g。下层氢氧化钠水溶液配置成30-50%水溶液可以循环使用于粗甲基氮丙啶的制备。
43.对本实施例所制备的2-甲基氮丙啶单体进行气相色谱纯度检测,检测结果如附图2和下表2所示。
44.表2
45.项目名称纯度/%水分/%精制甲基氮丙啶99.710.02
46.实施例3
47.粗氮丙啶制备:准确称取67g单乙醇胺,常温滴加98%浓盐酸108g,5h滴完,滴毕保温温度为80℃,保温2h,100℃、0.5mpa进行减压蒸馏,4h蒸完,降温至20℃,加入质量浓度为35%的236g氢氧化钠水溶液,升温40℃,反应2h,50℃、0.8mpa进行减压蒸馏3h,得到含量为45%氮丙啶水溶液118g。
48.精制氮丙啶制备:取含量为45%氮丙啶水溶液118g,加入61g氢氧化钠于温度为8℃的反应釜中,开动搅拌,使氢氧化钠溶液达到饱和状态,未饱和时继续加30g片碱,直至反应溶液达到饱和为止,搅拌2h,静置3h,静置温度为8℃,静置结束后将上层氮丙啶转移精馏釜,通氮气,进行精馏,精馏条件为:50℃,0.8mpa,精馏得到纯度大于99%的氮丙啶单体63g。下层氢氧化钠水溶液配置成30-50%水溶液可以循环使用于粗氮丙啶的制备。
49.对本实施例所制备的氮丙啶单体进行气相色谱纯度检测,检测结果如附图3和下表3所示。
50.表3
51.项目名称纯度/%水分/%精制氮丙啶99.530.01
52.对比例1
53.粗氮丙啶制备:准确称取67g单乙醇胺,常温滴加98%浓盐酸108g,5h滴完,滴毕保温温度为80℃,保温2h,100℃、0.5mpa进行减压蒸馏,4h蒸完,降温至20℃,加入质量浓度为
35%的276g氢氧化钠水溶液,升温40℃,反应2h,50℃、0.8mpa进行减压蒸馏3h,得到含量为40%氮丙啶水溶液138g。
54.精制氮丙啶制备:取含量为40%氮丙啶水溶液138g,进行一次减压精馏,条件为:温度50℃,0.8mpa,5h。
55.得到纯度大于90%的氮丙啶单体43g。
56.对本实施例所制备的氮丙啶单体进行气相色谱纯度检测,检测结果如附图4和下表4所示。
57.表4项目名称纯度/%水分/%精制氮丙啶90.72.20
58.通过对以上实施例和对比例,进行对比可以发现,使用本发明得到氮丙啶一次精馏得到的氮丙啶含量都大于99%,水分含量在0.01~0.02%,而不用本发明的方法处理,直接一次精馏所得到的氮丙啶的纯度只有90%左右,远远达不到精细化工使用要求,而且水分偏高。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1